
Enzyme replacement therapy for 15 years in Pompe disease: case report
Article Sidebar
Main Article Content
Abstract
Background: Pompe's disease or Glycogen Storage Disease type II (OMIM #232300), is an inherited metabolic disorder caused by a mutation in the GAA gene. This mutation affects the production of the acid α-glucosidase enzyme, leading to the accumulation of glycogen in the lysosome, especially in cardiac and skeletal muscle triggering multiple signs and symptoms of variable presentation.
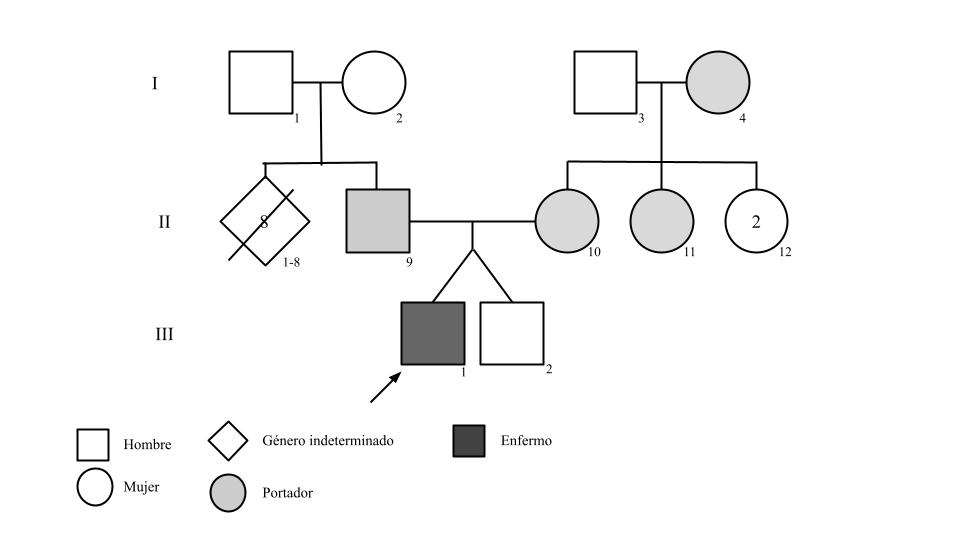
Case summary: The case presented is that of a 22-year old patient with a history of preterm birth and low weight, and a family history of GAA gene mutation in his parents who are carriers and twin brother who is not a carrier of this mutation. Beginning at 18 months old, the patient experienced difficulty feeding, facial muscle weakness, intermittent cyanosis, and progressive respiratory symptoms. He also presented with daily loose stools and liver abnormalities. After a series of adverse events, the diagnosis of glycogenosis was confirmed and treatment with enzyme replacement therapy was started, which resulted in improvement of symptoms. Currently, he remains stable with mild muscular and respiratory compromise.
Conclusions: This condition has a low prevalence and its presentation and severity vary. For its diagnosis, clinical suspicion and diagnostic tests such as enzymatic analysis and gene sequencing are required. Integral management involves several specialists and enzyme replacement therapy is the main treatment, although gene therapy shows promising advances.
Downloads
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Creative Commons
License Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0)
You are free to:
Share - copy and redistribute the material in any medium or format.
Adapt - remix, transform, and build upon the material The licensor cannot revoke these freedoms as long as you follow the license terms.
• Attribution — You must give appropriate credit, provide a link to the license, and indicate if changes were made. You may do so in any reasonable manner, but not in any way that suggests the licensor endorses you or your use.
• NonCommercial — You may not use the material for commercial purposes.
• ShareAlike — If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original.
• No additional restrictions — You may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.




References
- Sidorina A, Catesini G, Levi Mortera S, Marzano V, Putignani L, Boenzi S, et al. Combined proteomic and lipidomic studies in Pompe disease allow a better disease mechanism understanding. J Inherit Metab Dis. 2021 May;44(3):705-717. https://www.doi.org/10.1002/jimd.12344.
- omim.org. GLUCOSIDASE, ALPHA, ACID; GAA (*606800). https://www.omim.org/entry/606800. (Fecha de consulta: 17 de Abril, 2023)
- Marques JS. The Clinical Management of Pompe Disease: A Pediatric Perspective. Children. 2022 Sep 16;9(9):1404. https://www.doi.org/10.3390/children9091404
- Fatehi F, Ashrafi MR, Babaee M, Ansari B, Beiraghi Toosi M, Boostani R, et al. Recommendations for infantile-onset and late-onset Pompe disease: An Iranian consensus. Frontiers in Neurology. 2021;12. https://doi.org/10.3389/fneur.2021.739931
- Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child. 2020 Oct 2;24(2):3–8 https://doi.org/10.34763/jmotherandchild.20202402si.2001.000002
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Vol. 24, Current Treatment Options in Neurology. Springer; 2022. p. 573–88 .https://doi.org/10.1007/s11940-022-00736-1
- Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Vol. 15, Neurotherapeutics. Springer New York LLC; 2018. p. 928–42. https://doi.org/10.1007/s13311-018-0655-y.
- Taverna S, Cammarata G, Colomba P, Sciarrino S, Zizzo C, Francofonte D, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020 Aug 3;12(15):15856-15874. https://doi.org/10.18632/aging.103794
- Marisa Wexler MS. Pompe disease [Internet]. Pompe Disease News. 2022 [citado el 22 de abril de 2023]. Disponible en: https://pompediseasenews.com/what-is-pompe-disease/
- Unnisa Z, Yoon JK, Schindler JW, Mason C, van Til NP. Gene therapy developments for Pompe disease. Biomedicines [Internet]. 2022;10(2):302. Disponible en: http://dx.doi.org/10.3390/biomedicines10020302
- Sarah B, Giovanna B, Emanuela K, Nadi N, Josè V, Alberto P. Clinical efficacy of the enzyme replacement therapy in patients with late-onset Pompe disease: a systematic review and a meta-analysis. J Neurol [Internet]. 2022 [citado el 22 de abril de 2023];269(2):733–41. Disponible en: http://dx.doi.org/10.1007/s00415-021-10526-5
- Lecis M, Rossi K, Guerzoni ME, Mariotti I, Iughetti L. Enzyme replacement therapy (ERT) on heart function changes the outcome in patients with infantile-onset Pompe disease: A familial history. Case Rep Pediatr [Internet]. 2023 [citado el 23 de abril de 2023];2023:8470341. Disponible en: https://www.hindawi.com/journals/cripe/2023/8470341/
- Kohler L, Puertollano R, Raben N. Pompe disease: From basic science to therapy. Neurotherapeutics [Internet]. 2018 [citado el 23 de abril de 2023];15(4):928–42. Disponible en: https://pubmed.ncbi.nlm.nih.gov/30117059/
- Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child [Internet]. 2020;24(2):3–8. Disponible en: http://dx.doi.org/10.34763/jmotherandchild.20202402si.2001.000002
- Lecis M, Rossi K, Guerzoni ME, Mariotti I, Iughetti L. Enzyme replacement therapy (ERT) on heart function changes the outcome in patients with infantile-onset Pompe disease: A familial history. Case Rep Pediatr [Internet]. 2023 [citado el 23 de abril de 2023];2023:8470341. Disponible en: https://www.hindawi.com/journals/cripe/2023/8470341/