
Mucopolisacaridosis tipo I, variante síndrome de Hurler: Abordaje inicial y relación con la literatura
Barra lateral del artículo
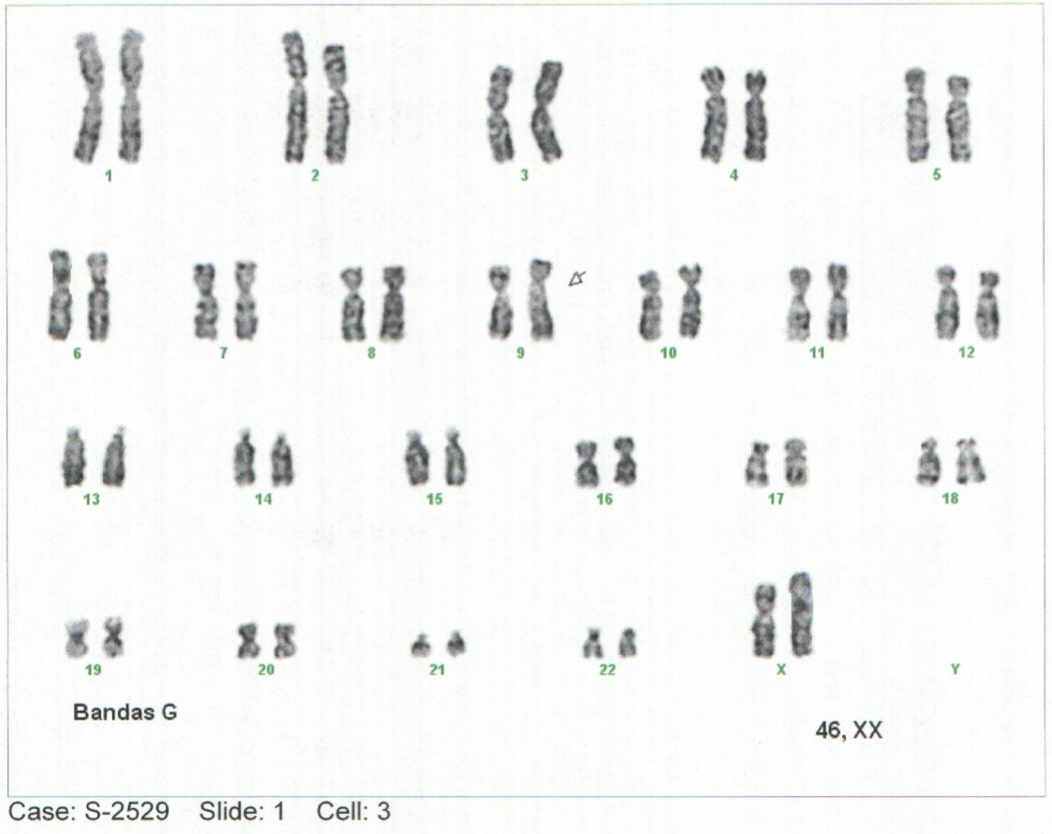
Contenido principal del artículo
Resumen
Antecedentes: las enfermedades de depósito tipo mucopolisacaridosis son un grupo de enfermedades genéticas poco frecuentes con patrón de herencia tipo autosómico recesivo. La mucopolisacaridosis (MPS) es considerada específicamente una condición de sobrecarga lisosomal causada por deficiencias de enzimas encargadas de la degradación de glicosaminoglicanos (GAG), también llamados mucopolisacáridos; este déficit enzimático se genera de la acumulación progresiva de compuestos en diferentes tejidos que conlleva a daño tisular generalizado y que tiende a progresar a falla multiorgánica (1–5). Reporte de caso: paciente femenina lactante mayor con retraso en el neurodesarrollo y alteraciones fenotípicas notorias, lo cual se relaciona con hallazgos descritos en la literatura. Conclusiones: se identificó déficit de la enzima alfa–L-iduronidasa, en contexto de un cuadro clínico con manifestaciones severas y la edad tan temprana de inicio de la patología, se cataloga dentro de la MPS I, Síndrome de Hurler. El avance en el abordaje temprano y conocimiento en la historia natural de las enfermedades de depósito permitirá generar un mejor abordaje diagnóstico y terapéutico, generando un mejor desenlace.
Descargas
Detalles del artículo

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-SinDerivadas 4.0.
Licencia Creative Commons
Atribución-NoComercial-CompartirIgual 4.0 Internacional (CC BY-NC-SA 4.0)
Usted es libre de:
Compartir - copiar y redistribuir el material en cualquier medio o formato.
Adaptar - remezclar, transformar y construir a partir del material
La licencia no puede revocar estas libertades en tanto se sigan los términos de la licencia.
- Atribución — Usted debe dar crédito de manera adecuada, brindar un enlace a la licencia, e indicar si se han efectuado cambios. Puede hacerlo en cualquier forma razonable, pero no de forma tal que sugiera que usted o su uso tienen el apoyo del licenciante.
- NoComercial — Usted no puede hacer uso del material con propósitos comerciales.
- CompartirIgual— Si remezcla, transforma o crea a partir del material, debe distribuir su contribución bajo la misma licencia del original.
- No hay restricciones adicionales — No puede aplicar términos legales ni medidas tecnológicas que restrinjan legalmente a otras a hacer cualquier uso permitido por la licencia.




Citas
Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008;3(1):1–7. DOI: https://doi.org/10.1186/1750-1172-3-24
Filocamo M, Tomanin R, Bertola F, Morrone A. Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know. Ital J Pediatr. 2018;44(Suppl 2):129. DOI: https://doi.org/10.1186/s13052-018-0553-2
Feillet F, Wiedemann A, Jeannesson E, Jaussaud R, Journeau P. Mucopolisacaridosis. EMC - Pediatría. 2016;51(3):1–14. DOI: https://doi.org/10.1016/S1245-1789(16)78912-8
Tebani A, Zanoutene-Cheriet L, Adjtoutah Z, Abily-Donval L, Brasse-Lagnel C, Laquerrière A, et al. Clinical and molecular characterization of patients with mucopolysaccharidosis type I in an Algerian series. Int J Mol Sci. 2016;17(5). DOI: https://doi.org/10.3390/ijms17050743
Khan SA, Peracha H, Ballhausen D, Wiesbauer A, Gautschi M, Mason RW, et al. Molecular Genetics and Metabolism. HHS Public Access Author. 2018;121(3):227–40. DOI: https://doi.org/10.1016/j.ymgme.2017.05.016
Spranger WJ, Kliegman R. Nelson. Tratado de pediatria - Mucopolisacaridosis [Internet]. 20th Editi. Nelson. Tratado de pediatría. Elsevier Espa8#241;a, S.L.U.; 2016. 772–779 p. Available from: https://www-clinicalkey-es.ez.unisabana.edu.co/#!/browse/book/3-s2.0-C20151024243
Simon Jones, MD, Robert Wynn M. Clinical features and diagnosis. UpToDate [Internet]. 2019; Available from: https://www-uptodate-com.ez.unisabana.edu.co/contents/mucopolysaccharidoses-clinical-features-and-diagnosis/print?search=mucopolisacaridosis&source=search_…
Lee-Chen GJ, Wang TR. Mucopolysaccharidosis type I: Identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J Med Genet. 1997;34(11):939–41. DOI: https://doi.org/10.1136/jmg.34.11.939
Yang C, Pan J, Linpeng S, Li Z, Tan H, Wu L. Identification of five novel mutations causing rare lysosomal storage diseases. Med Sci Monit. 2019;25:7634–44. DOI: https://doi.org/10.12659/MSM.915876
Scott HS, Bunge S, Gal A, Clarke LA, Phillip Morris C, Hopwood JJ, et al. Molecular Genetics of Mucopolysaccharidosis Type I: Diagnostic, Clinical, and Biological Implications. Hum Mutat. 1995;6:288302. DOI: https://doi.org/10.1002/humu.1380060403
Bianchi PM, Gaini R, Vitale S. ENT and mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):127. DOI: https://doi.org/10.1186/s13052-018-0555-0
Suarez-Guerrero JL, Gómez Higuera PJI, Arias Flórez JS, Contreras-García GA. Mucopolisacaridosis: características clínicas, diagnóstico y de manejo. Rev Chil Pediatr. 2016;87(4):295–304. DOI: https://doi.org/10.1016/j.rchipe.2015.10.004
Clarke LA, Atherton AM, Burton BK, Day-Salvatore DL, Kaplan P, Leslie ND, et al. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J Pediatr [Internet]. 2017;182:363–70. Available from: http://dx.doi.org/10.1016/j.jpeds.2016.11.036 DOI: https://doi.org/10.1016/j.jpeds.2016.11.036
Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12(1):1–10. DOI: https://doi.org/10.1186/s13023-017-0583-7
Harmatz PR, Shediac R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front Biosci - Landmark. 2017;22(3):385–406. DOI: https://doi.org/10.2741/4490
Coutinho MF, Encarnação M, Matos L, Silva L, Ribeiro D, Santos JI, et al. Molecular characterization of a novel splicing mutation underlying mucopolysaccharidosis (MPS) type VI—Indirect proof of principle on its pathogenicity. Diagnostics. 2020;10(2):1–11. DOI: https://doi.org/10.3390/diagnostics10020058
Peck DS, Lacey JM, White AL, Pino G, Studinski AL, Fisher R, et al. Incorporation of second-tier biomarker testing improves the specificity of newborn screening for mucopolysaccharidosis type i. Int J Neonatal Screen. 2020;6(1):1–10. DOI: https://doi.org/10.3390/ijns6010010
Shafaat M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic testing of Mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: in silico analysis of novel mutations. Metab Brain Dis. 2019;34(5):1447–55. DOI: https://doi.org/10.1007/s11011-019-00434-z
Li X, Xiao R, Chen B, Yang G, Zhang X, Fu Z, et al. A novel mutation of SGSH and clinical features analysis of mucopolysaccharidosis type IIIA. Med (United States). 2018;97(52). DOI: https://doi.org/10.1097/MD.0000000000013758
Parini R, Deodato F, Di Rocco M, Lanino E, Locatelli F, Messina C, et al. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12(1):1–9. DOI: https://doi.org/10.1186/s13023-017-0662-9
Sawamoto K, Chen HH, Alméciga-Díaz CJ, Mason RW, Tomatsu S. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018;123(2):59–68. DOI: https://doi.org/10.1016/j.ymgme.2017.12.434
Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019;2019(6). DOI: https://doi.org/10.1002/14651858.CD009354.pub5
Fecarotta S, Gasperini S, Parenti G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. Ital J Pediatr. 2018;44(Suppl 2):124. DOI: https://doi.org/10.1186/s13052-018-0564-z
Kuiper GA, Nijmeijer SCM, Roelofs MJM, van der Lee JH, Hollak CEM, Bosch AM. Limited data to evaluate real-world effectiveness of enzyme replacement therapy for mucopolysaccharidosis type I. J Inherit Metab Dis. 2019;42(5):762–75. DOI: https://doi.org/10.1002/jimd.12103
Eisengart JB, Rudser KD, Xue Y, Orchard P, Miller W, Lund T, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423–9. DOI: https://doi.org/10.1038/gim.2018.29