Pediatría
ISSN impreso: 0120-4912
e-ISSN: 2444-9369
DOI: 10.14295/rp.v57i3.513
Artículo de Investigación
Fernando Suarez Obando MD. Esp. MSca,b![]() , Álvaro Hernando Izquierdo Bello MD. Espc
, Álvaro Hernando Izquierdo Bello MD. Espc![]() , Juan Carlos Prieto Rivera MD. Esp.b,d
, Juan Carlos Prieto Rivera MD. Esp.b,d![]() , Fernando Ortiz MD. Esp.e,f
, Fernando Ortiz MD. Esp.e,f![]() , Melissa Díaz MD. MSc.g
, Melissa Díaz MD. MSc.g![]() , Kevin Maldonado MD. MSc.g
, Kevin Maldonado MD. MSc.g![]() , Carolina Baquero MD. Esph,i
, Carolina Baquero MD. Esph,i![]() , Edna Julieth Bobadilla MD. Esp. MSc.j,k
, Edna Julieth Bobadilla MD. Esp. MSc.j,k![]() , Sandra Milena Castellar MD. Esp.e,f,l
, Sandra Milena Castellar MD. Esp.e,f,l![]() , Edicson Ruiz MD. Esp.e,f,l, Sandra Catalina Mesa MD. Esp.h, Fabián Hernández BPharm. MSc.g
, Edicson Ruiz MD. Esp.e,f,l, Sandra Catalina Mesa MD. Esp.h, Fabián Hernández BPharm. MSc.g
a. Servicio de Genética, Instituto de Ortopedia Infantil Roosevelt, Bogotá, Colombia.
b. Instituto de Genética Humana, Facultad de Medicina, Pontificia Universidad Javeriana, Bogotá, Colombia.
c. Departamento de Pediatría, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, Colombia.
d. Servicio de Genética, Hospital La Victoria, Bogotá, Colombia.
e. Servicio de Fisiatría, Instituto de Ortopedia Infantil Roosevelt, Bogotá, Colombia.
f. Departamento de Medicina Física y Rehabilitación, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, Colombia.
g. Real World Insights (RWI), IQVIA, Bogotá, Colombia.
Financiación: Este manuscrito fue financiado por Biogen.
Fecha de recepción: 14 de diciembre de 2023; Fecha de aceptación: 26 de septiembre de 2024
Cómo citar: Suárez Obando F, Izquierdo Bello AH, Prieto Rivera JC, Ortiz F, Díaz M, Maldonado K, Baquero C, Bobadilla EJ, Castellar SM, Ruiz E, Mesa SC, Hernández F. Algoritmo de orientación para la identificación temprana de los signos, síntomas y el diagnóstico de AME 5q 1-3 en Colombia: Panel Delphi Modificado. Pediatr. 2024;57(3):e513.
Autor de correspondencia: Melissa Díaz MD. MSc. Correo electrónico: ;
Editor en jefe: Álvaro León Jácome Orozco
Resumen
Introducción: existe un vacío conceptual sobre la realización temprana de la confirmación genética para la Atrofia Muscular Espinal (AME). Se propone, por medio de un panel Delphi modificado, un algoritmo que oriente pragmáticamente al personal de salud, en la identificación temprana de los signos y síntomas de AME 5q tipos 1 a 3, con el fin de incrementar el rendimiento diagnóstico, clínico y molecular.
Materiales y Métodos: utilizando Delphi modificado, tres expertos con amplia experiencia clínica, investigativa y docente desarrollaron 13 casos, teniendo como insumo una revisión descriptiva de la literatura y su experiencia asistencial. Posteriormente, un grupo extendido de nueve expertos participó en dos rondas calificativas para cada caso utilizando la escala Likert.
Resultados: ocho expertos completaron la primera ronda, reportaron una mediana de ejercicio asistencial como especialista de 22.5 años (rango 5 a 35) y una mediana de pacientes con AME tratados de 50 (rango 15 a 150). En la primera ronda, se logró consenso en 19 afirmaciones para AME-1, 19 afirmaciones para AME-2 y 15 afirmaciones para AME-3. En la segunda ronda se logró consenso para todas las afirmaciones de examen físico y paraclínicos, mientras que las afirmaciones sin consenso se relacionaron a diagnósticos diferenciales. Hubo consenso en 72.7% de 11 afirmaciones para AME-1, en 85.7% de ocho afirmaciones para AME-2 y en 100% de ocho afirmaciones para AME-3.
Conclusiones: la metodología Delphi permitió configurar una aproximación algorítmica en forma de flujograma para la identificación temprana de los signos y síntomas y el diagnóstico de cada subtipo de AME 5q. Se propone un sustento integral y pragmático que sirva de orientación para el médico general y especialista, enfatizando en la importancia de su capacitación en la evaluación de signos y síntomas para considerar esta enfermedad tempranamente en el diagnostico diferencial.
Palabras clave: Atrofia Muscular Espinal, Diagnóstico, Consenso, América Latina, AME 5q.
Abstract
Introduction: There is a conceptual gap regarding early genetic confirmation for Spinal Muscular Atrophy (SMA). An algorithm is proposed, through a modified Delphi panel, to pragmatically guide healthcare personnel in the early identification of signs and symptoms of SMA types 1 to 3, aiming to increase diagnostic, clinical, and molecular performance.
Materials and Methods: Using a modified Delphi approach, a group of three experts with extensive clinical, research, and teaching experience developed 13 cases based on their clinical expertise and a descriptive literature review. Subsequently, an extended group of nine experts participated in 2 rating rounds for each case using the Likert scale.
Results: Eight experts completed the first round, reporting a median of 22.5 years of specialist clinical practice (range 5 to 35) and a median of 50 treated SMA patients (range 15 to 150). In the first round, consensus was reached on 19 statements for SMA-1, 19 statements for SMA-2, and 15 statements for SMA-3. In the second round, consensus was achieved in 72.7% of 11 statements for SMA-1, 85.7% of eight statements for SMA-2, and 100% of eight statements for SMA-3.
Conclusions: The Delphi methodology allowed for the development of an algorithmic approach in the form of a flowchart for the early identification of signs, symptoms, and diagnosis of each subtype of 5q SMA. A comprehensive and pragmatic framework is proposed to serve as guidance for general and specialist physicians.
Keywords: Spinal Muscular Atrophy, Diagnosis, Consensus, Latin America, 5q SMA.
La Atrofia Muscular Espinal (AME, MIM #253300) 5q es una enfermedad neurodegenerativa autosómica recesiva caracterizada por debilidad muscular severa secundaria a variantes patogénicas en el gen SMN1 (5q11.1). En la AME, la copia telomérica del gen SMN (SMNt, 5q13.2) generalmente carece de expresión debido a deleciones, siendo la más frecuente la deleción homocigota del exón 7 o, rara vez, a variantes patogénicas puntuales que interrumpen la función de la proteína de supervivencia de la neurona motora (SMN). La copia centromérica, altamente homóloga del SMN (SMNc, 5q13.2), contiene una sustitución tipo transición silenciosa (840C→T), cuyo producto proteico SMNΔ7 no es funcionalmente equivalente a SMN. Actualmente, SMNT se denomina SMN1 y SMNC se denomina SMN2 (1, 2). La AME se clasifica en 5 subtipos, AME 0 a 3 de la infancia y AME tipo 4 del adulto (3, 4).
Es una enfermedad rara con una incidencia global de 8 a 11.9 casos por 100,000 nacidos vivos (5, 6) y una prevalencia de 1 a 13 casos por 100,000 habitantes. Estas cifras varían según la ubicación geográfica y el método de detección (i.e., clínica vs. genética) (5). La AME-1 es la más frecuente (AME-1: 58-60%; AME-2: 20-29% y AME-3: 20-13%) (5, 7, 8), sin embargo, su prevalencia de 0.04 a 0.28 casos por 100,000 habitantes está condicionada por su mal pronóstico, en contraste a la de 1.5 casos por 100,000 habitantes de la AME 2 y la AME 3 combinadas (5).
En Latinoamérica los estudios son limitados, destacando el estudio en México con reporte de incidencia de 0.5-1 casos por 100 000 nacidos vivos (9). En Colombia, series de entre 14 a 29 casos han mostrado una predominancia de AME-2 (62-71.4%), con una supervivencia global de 28.6 años y tendencias de tiempo al diagnóstico similares entre subtipos (10, 11).
El espectro fenotípico de la AME incluye la neuropatía puramente motora, arreflexia y fasciculaciones, con sensibilidad conservada y un patrón típico de debilidad muscular de predominio proximal, principalmente de las extremidades inferiores, asociado a función cognitiva normal pero que con el tiempo puede comprometerse, en especial para AME-1 y 2 (12).
La AME-0 es de inicio antenatal, tiene un pronóstico muy pobre con expectativa de vida de semanas por falla ventilatoria. Puede ser sospechada dada la disminución de la movilidad del feto secundaria a hipotonía y debilidad muscular marcada (13), así como por signos indirectos de hipoquinesia fetal (e.g., polihidramnios, restricción del crecimiento intrauterino, contracturas articulares múltiples [artrogriposis] e hipoplasia pulmonar) (14). Se acompaña frecuentemente de arreflexia, defectos anatómicos cardiacos congénitos (e.g., comunicación interauricular e interventricular) (15).
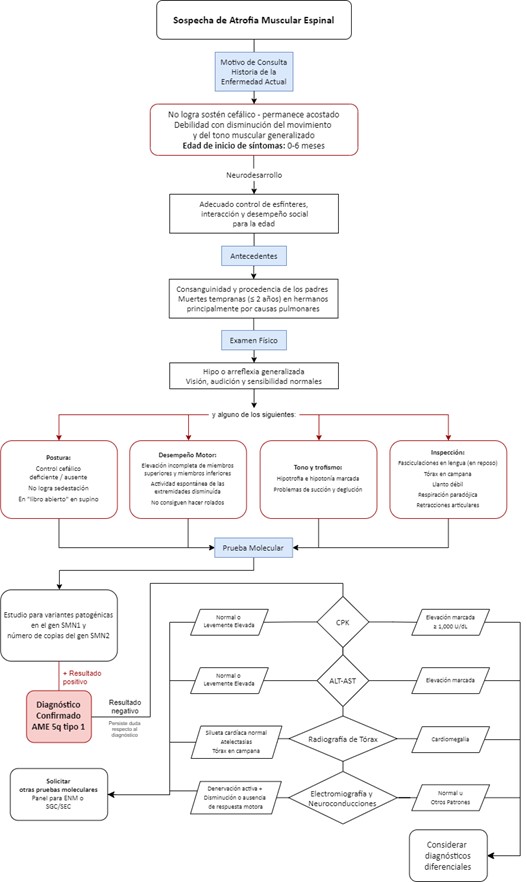
La AME-1 inicia de 0 a 6 meses, los pacientes no logran hacer rolados ni sentarse, por lo que se conocen de acuerdo a la máxima función motora alcanzada como “non-sitter” o “no sedentes”, predominando una postura en libro abierto en supino, arreflexia, hipotonía marcada, respiración paradójica, tórax acampanado, compromiso deglutorio, fasciculaciones en lengua y una sobrevida de aproximadamente 10 meses debido a complicaciones respiratorias (3, 4, 16).
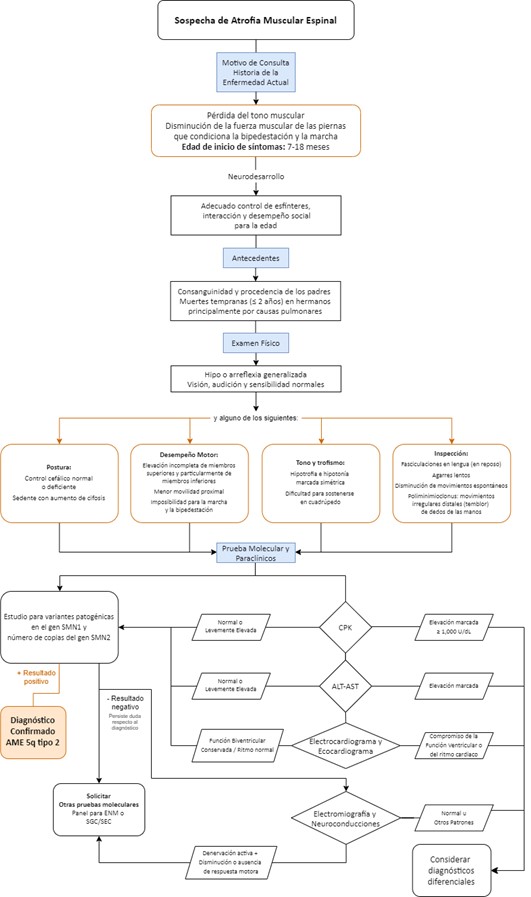
La AME-2 comienza de 7 a 18 meses. Puesto que los pacientes alcanzan el sedente, se conocen como “sitter” o “sedentes”, pero no son caminadores. Presentan hipotonía y debilidad predominantemente proximal y de miembros inferiores, debilidad de los músculos intercostales, arreflexia, escoliosis, poliminimioclonías (temblor fino en manos), contracturas articulares y enfermedad pulmonar de patrón restrictivo limitando la supervivencia hasta alrededor de los 20 años (3, 4, 17).
La AME-3 inicia entre los 1.5 y 10 años. Los pacientes caminan con limitaciones por debilidad proximal y de miembros inferiores. Se conocen como “walkers” o “caminadores”, sin compromiso pulmonar, logrando una supervivencia cercana a la población general (3, 4, 17).
El proceso diagnóstico comienza con la detección de signos y síntomas o mediante pruebas genéticas, las cuales pueden realizarse en tamizajes neonatales universales o en presencia de antecedentes familiares, remplazando así, la electromiografía y la biopsia muscular (4). El tratamiento integral incluye educación a familiares y cuidadores, rehabilitación muscular, uso de órtesis para apoyar la deambulación, prevención de complicaciones respiratorias manteniendo las vías aéreas despejadas con apoyo a la tos, soporte con ventilación no asistida nocturna y asistida nocturna o continua cuando necesario, y terapia deglutoria o soporte enteral en casos avanzados (18). Los tratamientos modificadores de la enfermedad que modulan la expresión del gen SMN son cruciales, especialmente si se inician temprano (19, 20), afirmando así la necesidad de confirmación temprana del caso.
Aunque existen guías terapéuticas para el manejo de AME, existe un vacío en la correlación entre signos y síntomas altamente sugestivos de la enfermedad con la necesidad de la realización temprana de la confirmación genética, especialmente en el contexto de recursos limitados para la atención en salud (10). Por ello, se hace necesario desarrollar un algoritmo que oriente pragmáticamente al personal de salud, con la identificación temprana de los signos y síntomas de AME tipos 1 a 3, con el fin de incrementar el diagnóstico, clínico y molecular, basado en una aproximación clínica sistemática ante la sospecha de AME.
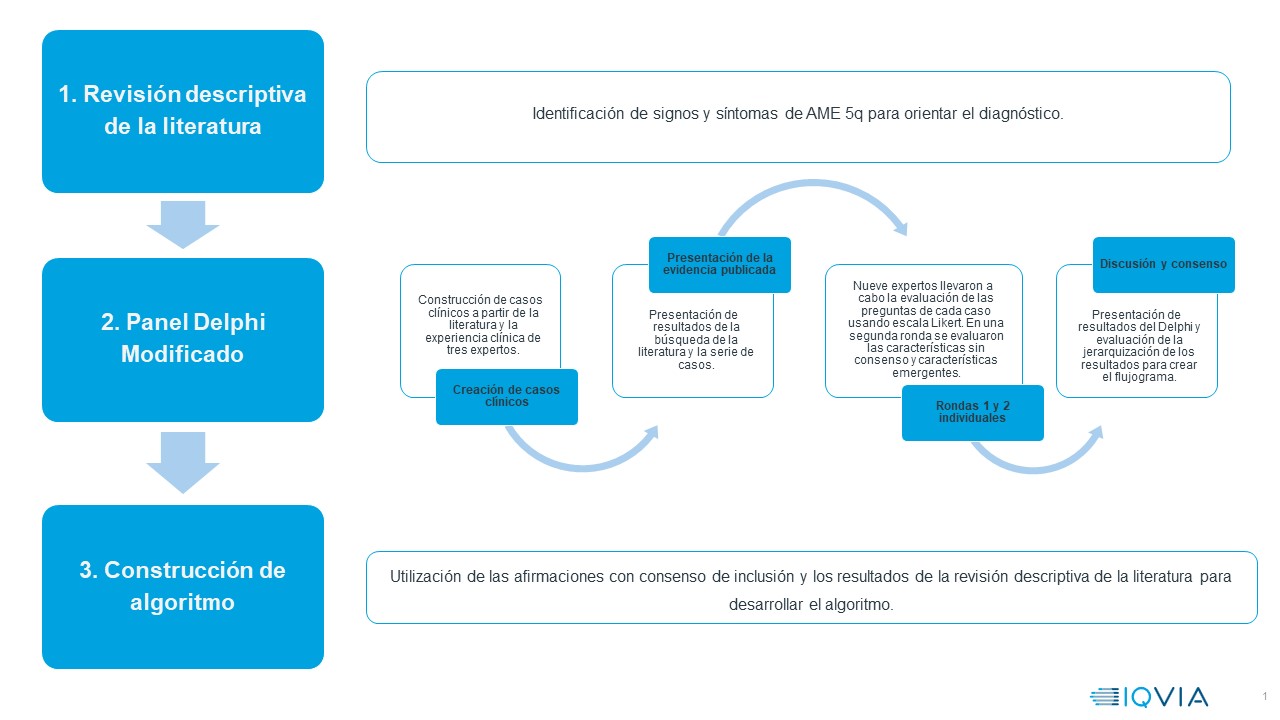
La metodología se desarrolló en tres etapas consecutivas (figura 1) que permitieron desarrollar un algoritmo de aproximación diagnóstica de AME tipos 1 a 3. A continuación, se explica cada etapa.
.
Figura 1. Metodología para el desarrollo de los algoritmos diagnósticos.Revisión descriptiva de la literatura sobre los signos y síntomas de pacientes con AME 5q para orientar el proceso diagnóstico (21). Se realizó una búsqueda estructurada en la base de datos MEDLINE utilizando los términos "spinal muscular atrophy, diagnosis, screening, physical examination, natural history” en diferentes combinaciones. La estrategia completa se encuentra en S1.
La metodología Delphi se emplea para identificar prioridades, alcanzar consensos y establecer directrices clínicas (22). En este estudio se empleó una metodología Delphi modificada (23-26), debido a su eficacia en contextos clínicos similares, donde la experiencia profesional acumulativa y la interdisciplinariedad son cruciales para asegurar diagnósticos precisos y tratamientos oportunos (23, 27-29). Se convocó a un grupo de tres expertos clínicos (AI, FO, JCP) de diferentes especialidades (neuropediatría, fisiatría y genética), con amplia experiencia asistencial, investigativa y docente. El grupo desarrolló 13 casos clínicos (AME-1: cinco casos, AME-2: cuatro casos y AME-3: cuatro casos). Los casos incluyeron signos y síntomas frecuentes, apoyos diagnósticos utilizados en la práctica y particularidades clínicas y paraclínicas que complican el diagnóstico diferencial.
Posteriormente, se invitó a un grupo extendido de expertos, usando como criterios de inclusión: mínimo 5 años de ejercicio como fisiatra, genetista o neuropediatra al cuidado de pacientes con AME, participación académica o autoría en publicaciones sobre AME y pertenecer a un centro de referencia de enfermedades neuromusculares nacional.
En la primera ronda, cada experto, a través de una encuesta anónima, calificó su grado de acuerdo con las situaciones clínicas presentadas. Se utilizó una escala Likert de 0 (completamente en desacuerdo) a 4 (completamente de acuerdo), con 2 indicando que no se está de acuerdo ni en desacuerdo. Mediante preguntas abiertas se indagó sobre otras características de interés para el clínico. El consenso se basó en la mediana: >2 para inclusión y <2 para exclusión. Aquellas con mediana igual a 2 fueron incluidas en la segunda ronda (23), donde se presentaron 3 casos clínicos, uno para cada subtipo de AME, enfocándose características sin consenso previo y temas emergentes de las preguntas abiertas. Los casos clínicos se encuentran en el material suplementario (Anexos Tabla S1 y S2).
Las afirmaciones con consenso de inclusión en ambas rondas y los resultados de la revisión descriptiva de la literatura fueron utilizadas para desarrollar un algoritmo de aproximación diagnóstica de AME5q subtipos 1-3 de forma individual-detallada y unificado-general. Las afirmaciones con consenso de exclusión fueron descartadas. La versión final se refinó con aportes del panel de expertos. Para ilustrar el algoritmo se empleó el software draw.io, versión diagrams.net 14.6.13.
.
.
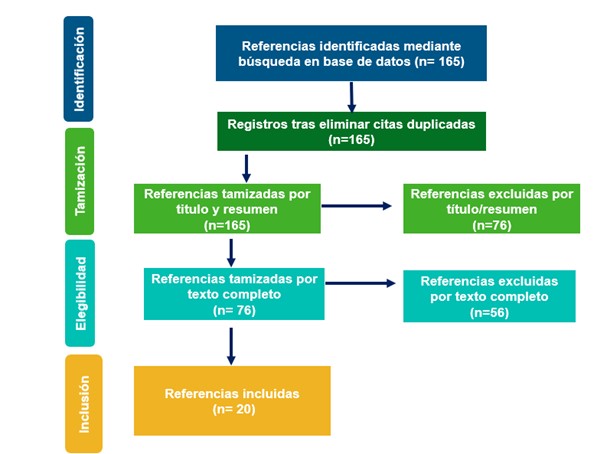
Figura 2. Selección de artículos de la revisión descriptiva de la literatura..
Posterior a eliminar duplicados y aplicar criterios de elegibilidad, se incluyeron 20 referencias en el análisis descriptivo (Ver Figura 2). Se encontraron 11 reportes de caso con diagnostico confirmado de AME-1 cuya edad al primer síntoma se encontraba entre 15 días y 5 meses (30-33). Solo en un caso se reportó antecedente familiar de AME. El síntoma por el que consultaban con mayor frecuencia fue hipotonía (55%) y debilidad muscular (46%). Al examen físico se confirmaba hipotonía en un 82% de los pacientes, debilidad muscular en el 64%, retraso o incapacidad para el sostén cefálico en el 46%, fasciculaciones en la lengua en el 46%, arreflexia o hiporreflexia 46%, 36% infecciones respiratorias recurrentes, llanto débil en el 23%, y en el 18% trastorno de la deglución, respiración paradójica o tórax en campana. En un paciente se encontró postura en libro abierto.
Para AME-2 se encontraron dos reportes de casos confirmados con edad del primer síntoma en promedio de 11 meses. Los síntomas presentados inicialmente fueron debilidad y disminución de los movimientos, en los dos casos se encontró retraso en el desarrollo motor, hipo o arreflexia, y no lograron marcha; en uno de los pacientes se reportó temblor en las manos y debilidad muscular proximal.
Por último, en 8 reportes de casos se reportó diagnóstico confirmado de AME-3 la edad promedio del primer síntoma fue de 9.8 años presentándose entre los 2 y 15 años, el 25% tenían antecedente familiar de AME y el 33% eran de sexo femenino. El 50% consultaron por debilidad muscular, 40% por problemas para caminar y 10% por caídas frecuentes. En todos los casos se confirmó debilidad muscular proximal, el 75% presentaba alteración o dificultad en la marcha, 63% hipo o arreflexia, 50% presentó signo de Gowers, 50% presentaba dificultad para subir escaleras. El 38% presentaba temblor descrito como temblor en los dedos de alta frecuencia y 25% dificultad para saltar.
Se convocó un panel de nueve expertos clínicos, tres de cada especialidad. Los ocho expertos que completaron la primera ronda tienen una mediana de tiempo de ejercicio asistencial como especialista de 22.5 años (rango 5 a 35 años) y una mediana de número de pacientes con AME tratados de 50 pacientes (rango 15 a 150). Adicionalmente, reportaron haber visto un total de 56 pacientes con AME-1, 83 pacientes con AME-2 y 37 pacientes con AME-3. En relación con la frecuencia de pacientes evaluados en el último año, AME se sospechó en 15, 17 y 11 casos de AME-1, 2 y 3 respectivamente, confirmando el diagnóstico en el 100, 53 y 64% de las ocasiones respectivas.
Se presentan los resultados de la primera ronda en la Tabla 1.
|
Contexto |
AME |
Afirmación |
CI |
CE |
2R |
||
|
1 |
2 |
3 |
|||||
|
Anamnesis |
|
|
|
Antecedentes
de consanguinidad en los padres |
|
|
|
|
|
|
|
Alteraciones
en la deglución |
|
|
|
|
|
Examen Físico |
|
|
|
Problemas
de succión |
|
|
|
|
|
|
|
Sostén
cefálico |
|
|
|
|
|
|
|
|
Llanto
débil |
|
|
|
|
|
|
|
|
Tórax
en campana |
|
|
|
|
|
|
|
|
Sensibilidad
conservada |
|
|
|
|
|
|
|
|
Disminución
de los movimientos espontáneos |
|
|
|
|
|
|
|
|
Fasciculaciones
de la lengua en reposo |
|
|
|
|
|
|
|
|
Debilidad
muscular de predominio proximal |
|
|
|
|
|
|
|
|
Atrofia
muscular proximal simétrica |
|
|
|
|
|
|
|
|
Cifosis |
|
|
|
|
|
|
|
|
Hipotonía |
|
|
|
|
|
|
|
|
Adecuada
sensibilidad |
|
|
|
|
|
|
|
|
Adecuado
intelecto, interacción social y estado de conciencia |
|
|
|
|
|
|
|
|
Adecuado
control de esfínteres |
|
|
|
|
|
|
|
|
Arreflexia
o hiporreflexia |
|
|
|
|
|
|
|
|
Debilidad
muscular |
|
|
|
|
|
|
|
|
Patrón
de marcha |
|
|
|
|
|
|
|
|
Coordinación
motora |
|
|
|
|
|
|
|
|
Movilidad
activa y pasiva de los tobillos |
|
|
|
|
|
|
|
|
Retracciones
articulares |
|
|
|
|
|
|
|
|
Alteraciones
en la deglución |
|
|
|
|
|
|
|
|
Soplos
cardiacos |
|
|
|
|
|
|
|
|
Respiración
paradójica |
|
|
|
|
|
|
|
|
Alteraciones
del lenguaje |
|
|
|
|
|
|
|
|
Alteraciones
del desarrollo cognitivo |
|
|
|
|
|
|
|
|
Fasciculaciones
en las extremidades |
|
|
|
|
|
|
|
|
Pseudohipertrofia
de gemelos |
|
|
|
|
|
|
|
|
Signo
de Gowers |
|
|
|
|
|
Paraclínicos |
|
|
|
Estudio
molecular del gen SMN1 |
|
|
|
|
|
|
|
CPK |
|
|
|
|
|
|
|
|
ALT-AST |
|
|
|
|
|
|
|
|
Electromiografías |
|
|
|
|
|
|
|
|
Neuroconducciones |
|
|
|
|
|
|
|
* |
Radiografía
de columna total |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
|
|
|
Biopsia
muscular |
|
|
|
|
|
|
|
|
Prueba
de 6 minutos** |
|
|
|
|
|
|
|
|
CHOP-INTEND |
|
|
|
|
|
|
|
|
Radiografía
de tórax |
|
|
|
|
|
|
|
|
Ecocardiograma |
|
|
|
|
|
|
|
|
Test
de estímulo repetitivo |
|
|
|
|
|
|
|
|
Relación
lactato piruvato |
|
|
|
|
|
|
|
|
Ecografía
abdominal |
|
|
|
|
|
|
|
|
Resonancia
magnética corporal de músculo |
|
|
|
|
Para los casos de AME-1, se alcanzó consenso en 19 afirmaciones referentes a la anamnesis, el examen físico y los paraclínicos complementarios. Tres afirmaciones de este conjunto de categorías se excluyeron.
Como condiciones particulares, se acordó que, en un paciente con llanto y succión normal, pero en el que se identifique arreflexia generalizada y actividad espontanea de las extremidades disminuida sin observarse fasciculaciones en lengua (Caso 4), se deben solicitar estudios de líquido cefalorraquídeo. Referente a los diagnósticos diferenciales, hubo consenso para los casos 1, 2 y 4 en que el más probable era AME-1. Pero en el caso 2, ante la posibilidad de considerar la enfermedad de Pompe como diferencial, el caso pasó a segunda ronda.
En el caso 3, se descartó el diagnóstico de AME-1 o PSM y por consenso se consideró que fuese enfermedad de Pompe. En el caso 4 se descartó el síndrome de Guillain Barre (SGB) y se incluyó como posibilidad la AME y la distrofia muscular congénita (DMC). En el caso 5, se descartó el diagnóstico de AME-1, polineuropatía motora axonal y el SGB, encontrando consenso de inclusión en considerar el diagnóstico de enfermedad de Pompe. Finalmente, pasó a segunda ronda el considerar como diagnósticos diferenciales las miopatías congénitas (MC), la distrofia muscular merosina negativa o una miopatía mitocondrial (MM).
Para los casos clínicos de AME-2, se alcanzó consenso en 19 afirmaciones referentes a la anamnesis, el examen físico y los paraclínicos complementarios, descartando una afirmación.
Referente a los probables diagnósticos diferenciales, hubo consenso en enunciar para los casos 6 y 7 que el diagnóstico más probable era AME-2. Para el caso 6 se descartó la distrofia muscular de Duchenne (DMD) y en el caso 7 se incluyó como diagnóstico diferencial la enfermedad de Pompe. En el caso 8, formulado como diagnóstico diferencial, el panel descartó AME-2 e incluyó en cambio la DMD como diagnóstico junto con la indicación de toma de prueba molecular para el gen de la distrofina.
A partir de las preguntas abiertas se incluyó en la segunda ronda el buscar en el examen físico poliminimioclonus, organomegalias, retracciones articulares y soplos cardiacos, así como la indicación de resonancia nuclear magnética (RNM) de columna total contrastada y ecocardiograma y el considerar como diagnósticos diferenciales la PSM, una MC o la DMC.
Para los casos clínicos de AME-3, se alcanzó consenso en 15 afirmaciones referentes a la anamnesis, el examen físico y los paraclínicos complementarios, excluyendo 2 afirmaciones.
La solicitud de radiografía de columna total se excluyó para el caso 11. Referente a los posibles diagnósticos diferenciales hubo consenso de inclusión en enunciar para los casos 10 y 11 la AME-3. Para el caso 10, pasó a segunda ronda el considerar que fuese una marcha en punta de pies idiopática; se alcanzó consenso de inclusión para la DMD y se descartó una polineuropatía motora axonal (PMA), adicionalmente, no se logró consenso en proponer como diagnóstico más probable la AME-3.
En la parte A del caso 11 hubo consenso en tener como diagnóstico diferencial un trastorno en el desarrollo de la coordinación y en la parte B una distrofia muscular de Duchenne/Becker (DMD/B); adicionalmente pasó a segunda ronda la posibilidad de miastenia gravis. Para el caso 12, hubo consenso de inclusión en dar como diagnóstico una distrofia muscular cintura-miembro, pese a que no fue formulado originalmente como diagnóstico diferencial; en la parte B hubo consenso de inclusión en la necesidad de solicitar un panel molecular para distrofias musculares y pasó a segunda ronda la posibilidad diagnostica de AME-3; finalmente, en la parte C del caso se descartó como diagnóstico diferencial la DMD/B y se incluyó AME-3, si se tenía el resultado negativo del panel molecular para distrofia muscular. El caso 13, formulado como caso de diagnóstico diferencial, descartó como diagnóstico AME-3, incluyendo como más probable una DMD, al encontrar signos de enfermedad de fibra muscular en la electromiografía y se consideró como diagnóstico diferencial la MC.
En la segunda ronda se propuso un caso clínico para cada subtipo de AME y se contó con la participación de 7 expertos. Se presentan los resultados en la Tabla 2.
.
|
Contexto |
AME |
Afirmación |
CI |
CE |
||
|
1 |
2 |
3 |
||||
|
Examen Físico |
|
|
|
Alteraciones en la deglución |
|
|
|
|
|
|
Posición
y movilidad de las caderas* |
|
|
|
|
|
|
|
Retracciones articulares |
|
|
|
|
|
|
|
Soplos cardiacos |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Respiración paradójica |
|
|
|
|
|
|
|
Poliminimioclonus, |
|
|
|
|
|
|
|
Organomegalias |
|
|
|
|
|
|
|
Alteraciones del lenguaje |
|
|
|
|
|
|
|
Alteraciones del desarrollo cognitivo |
|
|
|
|
|
|
|
Fasciculaciones en las extremidades |
|
|
|
|
|
|
|
Pseudohipertrofia de gemelos |
|
|
|
|
|
|
|
Signo de Gowers |
|
|
|
|
Paraclínicos |
|
|
|
Radiografía de tórax |
|
|
|
|
|
|
Ecocardiograma |
|
|
|
|
|
|
|
Biopsia muscular |
|
|
|
|
|
|
|
Test de estímulo
repetitivo |
|
|
|
|
|
|
|
Relación lactato piruvato |
|
|
|
|
|
|
|
Ecografía abdominal |
|
|
|
|
|
|
|
Resonancia
magnética de columna total |
|
|
|
|
|
|
|
Resonancia
magnética corporal de músculo |
|
|
|
AME: Atrofia muscular espinal; CI: Consenso de inclusión; CE: Consenso de exclusión.
Para todos los subtipos de AME se logró consenso en las afirmaciones de las categorías examen físico y paraclínicos. Para AME-1 hubo consenso de inclusión en que el caso correspondía a AME-1 y en descartar que correspondiera a una enfermedad de Pompe; sin embargo, no se logró consenso en cuanto al incluir como diagnósticos diferenciales las MC, la distrofia muscular merosina negativa o una MM. Por otro lado, para AME-2 hubo consenso de inclusión en considerar AME-2 como el diagnóstico más probable, y se descartó que correspondiese a una distrofia muscular congénita. Otros diagnósticos diferenciales incluidos por consenso fueron la PSM progresiva y la MC. Finalmente, para el caso de AME-3 hubo consenso en proponer AME-3, así como distrofia muscular cintura-miembro como posibles diagnósticos diferenciales y en descartar la miastenia gravis o una MC.
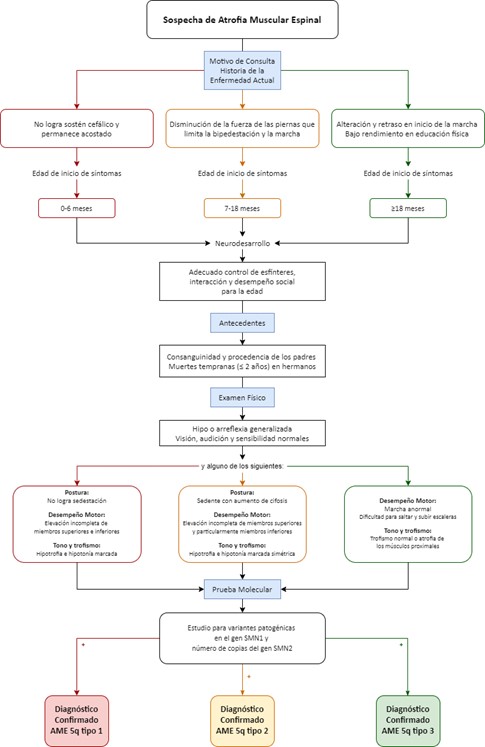
Flujograma para la identificación temprana de los signos y síntomas y confirmación diagnóstica de AME
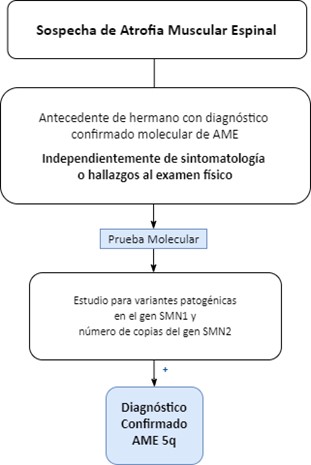
El algoritmo sigue una secuencia lógica, de “paso a paso” con progresión de decisiones, desde baja complejidad (primeros pasos), hasta a la mayor complejidad en los pasos finales. Los componentes principales del algoritmo se presentan en tres núcleos: 1. Signos y síntomas para identificación temprana, 2. Pruebas moleculares ante la sospecha diagnóstica y 3. Pruebas adicionales en caso en que la prueba molecular sea negativa y persista la sospecha diagnóstica. Se presenta en forma de flujograma una aproximación algorítmica unificada-general (Figura 3). Adicionalmente, se encuentra un flujograma para cuando el paciente cuente con antecedente de hermano(s) con diagnóstico de AME (Figura 4) y, como material suplementario, un flujograma detallado para AME-1, AME-2 y AME-3 (Figuras S1-S3).
.
.
Figura 3. Flujograma para la identificación temprana de los signos y síntomas y confirmación diagnóstica de AME 5q..
.
Figura 4. Flujograma para la identificación temprana y confirmación diagnóstica de AME 5q en pacientes con antecedente de hermano(s) con diagnóstico de AME.El panel Delphi modificado permitió, desarrollar una aproximación algorítmica en forma de flujograma para la identificación temprana de los signos y síntomas y el diagnóstico de cada subtipo de AME 5q.
Los resultados de la revisión de la literatura sirvieron como insumo para el desarrollo de casos por parte del grupo de 3 expertos clínicos, a su vez, dicho conjunto de casos, junto con la evaluación de consenso, sirvieron para el desarrollo del algoritmo teniendo como punto de partida el motivo de consulta del paciente. El algoritmo coincide con los ejercicios de anamnesis y examen físico que se hacen en cada consulta, los cuales delimitan los términos de solicitud de paraclínicos de diagnóstico o para enfocar el racionamiento asistencial hacia otros diagnósticos diferenciales, permitiendo al médico general o al especialista organizar su ejercicio clínico de forma integral y pragmática.
Dentro de las consideraciones específicas que brinden contexto al algoritmo, destaca en primer lugar el hecho de que la Electromiografía y Neuroconducciones ha de ser realizada por un médico especialista con experiencia en enfermedades neuromusculares, pues sus resultados son observador-dependientes. En segundo lugar, se hace énfasis en la necesidad de sospechar e identificar oportunamente la enfermedad en caso de encontrarse con un paciente quien tenga antecedente de hermano(s) con diagnóstico confirmado (molecular) de AME.
Independientemente de su sintomatología o de los hallazgos al examen físico se debe tomar una prueba molecular.
Una posible limitación del estudio es que entre los expertos hay diferencias en la frecuencia de años de ejercicio como especialista y en la cantidad de pacientes evaluados, sin embargo, es importante resaltar que estos aspectos están condicionados por la baja prevalencia de la AME.
El desarrollo del algoritmo se complementó con el sustrato teórico de la revisión de la literatura, y, al considerar como criterio de selección de los expertos, su participación en iniciativas académicas y eventos de divulgación científica local e internacional sobre AME, se concluye que es un grupo homogéneo y versado en la enfermedad. A lo anterior se suma que los expertos ejercen en centros de referencia nacional para la atención de enfermedades neuromusculares.
Respecto a la heterogeneidad de la condición, se buscó que en los casos clínicos de la primera ronda se abordaran los signos y síntomas más representativos presentando tanto “casos clásicos” como casos complejos pero con hallazgos puntuales reconocidos como propios de la condición, además de casos propuestos como diagnósticos diferenciales; Además, se preguntó a los expertos de forma abierta por aquellos diagnósticos que no hubiesen sido inicialmente incluidos llevándose estas propuestas para evaluación en la segunda ronda.
Al aplicar lo presentado al reporte de un caso publicado por Pane, 2017 (34) se encontró un paciente de cuatro meses que consultó por hipotonía y debilidad muscular cumpliendo con lo señalado por el algoritmo en donde no reportan alteración del control de esfínteres ni alteración del desarrollo del lenguaje. Al interrogar los antecedentes familiares, de acuerdo con el caso, no hay antecedentes. Continuando se encontraría en primer lugar un paciente con arreflexia y consecuentemente se haría necesario evaluar: a) postura, reportada como ausencia de control cefálico, b) desempeño motor, reportado como: algunos movimientos distales preservados de extremidades superiores con limitación de movimientos anti gravitacionales distales de las extremidades inferiores y debilidad severa, c) tono y trofismo, reportadas como hipotonía severa; cumpliendo así con los criterios para continuar la búsqueda de AME. De forma complementaria a la inspección en el examen físico se encontraría: tórax en campana, fasciculaciones en la lengua, problemas de deglución, respiración paradójica adicional al hallazgo previos de no lograr el sostén cefálico. En el caso reportado, se decidió solicitar la prueba RFLP-PCR para SMN1 exón 7 y 8 y el Número de copias del gen SMN2, lo que mostró deleción homocigota para el gen SMN1 y 2 copias de SMN2 confirmando el diagnóstico. Este ejercicio se propone con la intención de orientar al lector acerca de cómo aplicar el algoritmo, no siendo un método para su validación.
Se presenta una aproximación algorítmica en forma de flujograma para uso del médico general y del especialista no experimentado en AME 5q (tipos 1, 2 y 3). Se invita a tener especial atención en el hecho de que los paraclínicos complementarios nunca deben retrasar o reemplazar la indicación de la prueba molecular; de la misma manera, se debe recordar la importancia del diagnóstico temprano, ya que en especial para AME-1, un tratamiento oportuno puede cambiar significativamente el pronóstico del paciente. Para esto, se destaca la importancia de capacitar a los profesionales de la salud para una adecuada evaluación de los síntomas y signos, considerando tempranamente esta enfermedad como parte de los diagnósticos diferenciales clínicos.
Lo propuesto ha de ser considerado como una orientación que, en conjunto con el juicio clínico y el grueso de la literatura disponible al respecto, cumpla con el ideal de identificar, sospechar y diagnosticar de forma oportuna los casos de AME. Se propone como próximos pasos su validación en la práctica con intención de establecer su desempeño.
El apoyo de redacción para la preparación de este manuscrito fue proporcionado por IQVIA y financiado por Biogen. Los expertos clínicos que participaron en el panel Delphi recibieron una compensación económica por su tiempo. FO ha recibido honorarios como speaker de Biogen. Los autores no reportan otras afiliaciones relevantes o participación financiera con ninguna organización o entidad con un interés o un conflicto con el tema o los materiales discutidos en el manuscrito además de los divulgados.
1. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307-11.
2. Bürglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C, et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. 1996;32(3):479-82.
3. Darras BT. Spinal muscular atrophies. Pediatr Clin North Am. 2015;62(3):743-66.
4. Mercuri E, Sumner CJ, Muntoni F, Darras BT, Finkel RS. Spinal muscular atrophy. Nat Rev Dis Primers. 2022;8(1):52.
5. Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124.
6. Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27-32.
7. Verhaart IEC, Robertson A, Leary R, McMacken G, König K, Kirschner J, et al. A multi-source approach to determine SMA incidence and research ready population. J Neurol. 2017;264(7):1465-73.
8. Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet. 2004;12(12):1015-23.
9. Urrutia-Osorio ME, Ruiz-García M. Demographic and clinical profile in patients with spinal muscular atrophy: Series of 31 patients. Acta Pediátrica de México. 2020;41(2):47-57.
10. Cardona N, Ocampo SJ, Estrada JM, Mojica MI, Porras GL. Caracterización clínica y funcional de pacientes con atrofia muscular espinal en el centro-occidente colombiano. Biomédica. 2022;42(Sp. 1):89-99.
11. Valencia HD, Rendón Muñoz J, Pineda N, Ortiz B, Montoya JH, Cornejo JW. Características clínicas de los pacientes menores de 18 años con atrofia muscular espinal en Medellín, 2008-2013. Acta Neurológica Colombiana. 2016;32(1):9-17.
12. Masson R, Brusa C, Scoto M, Baranello G. Brain, cognition, and language development in spinal muscular atrophy type 1: a scoping review. Dev Med Child Neurol. 2021;63(5):527-36.
13. Grotto S, Cuisset JM, Marret S, Drunat S, Faure P, Audebert-Bellanger S, et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J Neuromuscul Dis. 2016;3(4):487-95.
14. González De Dios J, Martínez Frías ML, Arroyo Carrera I, Fondevilla Saucí J, Sanchís Calvo A, Hernández Ramón F, et al. [Role of signs of fetal hypokinesia in the diagnosis of spinal muscular atrophy of neonatal onset]. An Esp Pediatr. 2002;56(3):233-40.
15. Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45(10):635-8.
16. Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157-67.
17. Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67-72.
18. Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-15.
19. Reilly A, Chehade L, Kothary R. Curing SMA: Are we there yet? Gene Ther. 2023;30(1-2):8-17.
20. Mercuri E, Pera MC, Scoto M, Finkel R, Muntoni F. Spinal muscular atrophy - insights and challenges in the treatment era. Nat Rev Neurol. 2020;16(12):706-15.
21. Paré G, Trudel M-C, Jaana M, Kitsiou S. Synthesizing information systems knowledge: A typology of literature reviews. Information & Management. 2015;52(2):183-99.
22. Shang Z. Use of Delphi in health sciences research: A narrative review. Medicine. 2023;102(7):e32829.
23. Röth A, Maciejewski J, Nishimura JI, Jain D, Weitz JI. Screening and diagnostic clinical algorithm for paroxysmal nocturnal hemoglobinuria: Expert consensus. Eur J Haematol. 2018;101(1):3-11.
24. Eubank BH, Mohtadi NG, Lafave MR, Wiley JP, Bois AJ, Boorman RS, Sheps DM. Using the modified Delphi method to establish clinical consensus for the diagnosis and treatment of patients with rotator cuff pathology. BMC Med Res Methodol. 2016;16:56.
25. Dalkey NC. The Delphi method: An experimental study of group opinion. RAND CORP SANTA MONICA CA; 1969.
26. Dalkey N, Helmer O. An experimental application of the Delphi method to the use of experts. Management science. 1963;9(3):458-67.
27. Kaliner MA, Amin AN, Gehling R, Crivera C, Chiao E, Vishalpura T, Bramley TJ, editors. Impact of Inhaled Corticosteroid-Induced Oropharyngeal Adverse Effects on Treatment Patterns and Costs in Asthmatic Patients: Results from a Delphi Panel2005.
28. St Louis EK, Gidal BE, Henry TR, Kaydanova Y, Krumholz A, McCabe PH, et al. Conversions between monotherapies in epilepsy: expert consensus. Epilepsy Behav. 2007;11(2):222-34.
29. Strupp J, Romotzky V, Galushko M, Golla H, Voltz R. Palliative care for severely affected patients with multiple sclerosis: when and why? Results of a Delphi survey of health care professionals. J Palliat Med. 2014;17(10):1128-36.
30. Anagnostou E, Miller SP, Guiot MC, Karpati G, Simard L, Dilenge ME, Shevell MI. Type I spinal muscular atrophy can mimic sensory-motor axonal neuropathy. J Child Neurol. 2005;20(2):147-50.
31. da Silva LR, Colovati ME, Coprerski B, de Andrade CE, Zanoteli E, Raskin S, et al. Spinal muscular atrophy due to a "de novo" 1.3 Mb deletion: implication for genetic counseling. Neuromuscul Disord. 2013;23(5):388-90.
32. Eggermann T, Eggermann K, Elbracht M, Zerres K, Rudnik-Schöneborn S. A new splice site mutation in the SMN1 gene causes discrepant results in SMN1 deletion screening approaches. Neuromuscul Disord. 2008;18(2):146-9.
33. Kawashima H, Ishii C, Yamanaka G, Ioi H, Nishimata S, Kashiwagi Y, et al. Myopathy and neurogenic muscular atrophy in unexpected cardiopulmonary arrest. Pediatr Int. 2011;53(2):159-61.
34. Pane M, Lapenta L, Abiusi E, de Sanctis R, Luigetti M, Palermo C, et al. Longitudinal assessments in discordant twins with SMA. Neuromuscul Disord. 2017;27(10):890-3.
S1. Estrategia de búsqueda de la literatura
("screening"[ ("spinal muscular atrophy"[Title/Abstract] and (("diagnosis" [Title/Abstract]) or ("screening"[Title/Abstract]) or ("physical examination"[Title/Abstract]) or ("natural history"[Title/Abstract]) ) con los filtros “Full text, Case Reports, Guideline, Meta-Analysis, Practice Guideline, Review, Systematic Review, Humans, English, Spanish, only older than new borns”.
|
Caso
clínico |
Descripción |
|
AME 1 |
|
|
1 |
Parte A: Paciente de 6 meses femenina es traída
a consulta por sus padres porque aún no sostiene bien la cabeza, esta
acostada y poco se mueve. La mama la siente como una gelatina. Sin embargo,
tiene adecuada interacción con el medio. Antecedente familiar: retardo mental
tío abuelo. Parte B: Examen físico: Ella no sube-eleva los
miembros inferiores, no hay adecuado control cefálico. No hay disminución del
volumen muscular. No se atora cuando come, Dice ma-ma,
pa-pa. Sensibilidad normal. No hay deformidades. Se
observa arreflexia. Ecografía cerebral normal, y audición normal. |
|
2 |
Parte A: Paciente de 5 meses femenina es traída
a consulta por sus padres porque la siente muy débil y comparada a su
hermanito se mueve menos. Antecedente familiar: Los papas son primos en
tercer grado. Parte B: Examen físico: No se observa
alteración de la sensibilidad. Se observa problemas en la succión. Ella no
sube-eleva los miembros inferiores, no hay adecuado control cefálico. Se
observa hiporreflexia. Hay presencia de llanto débil. |
|
3 |
Parte A: Paciente de 6 meses masculino es
traído a consulta por sus padres porque no sostiene la cabeza y comparado a
su hermanita se mueve menos. Antecedente familiar: sin antecedentes de
importancia. Parte B: Examen físico: No se observa
alteración de la sensibilidad. No sube-eleva los miembros inferiores, no hay
adecuado control cefálico. Reflejos osteotendinosos normales. Disminución del
volumen muscular. CPK en 10,000 u/dL ALT-AST
elevadas. |
|
4 |
Parte A: Paciente de 2 meses, masculino, traído
por su madre quien refiere que el niño empezó a perder la movilidad de sus 4
extremidades desde hace 5 días.
Control de esfínteres normales. Antecedente familiar: un hermano del
niño falleció a los 2 años por neumonía. Parte B: Examen físico: se observa llanto y succión normal. Se
encuentra arreflexia generalizada. La actividad espontanea de las
extremidades está disminuida. No se observan fasciculaciones en lengua. Parte C: Se recibe el resultado de la CPK:
1,000 u/dL. Parte D: Se recibe el resultado de: Neuroconducciones: ausencia de respuesta motora de los
nervios medianos y del nervio tibial derecho. El nervio tibial izquierdo
tiene una amplitud de 0,9 mV. Las neuroconducciones sensitivas están normales.
Electromiografía: se encuentran signos de denervación activa, unidades de
gran amplitud y larga duración en el músculo iliopsoas derecho. |
|
5 (Diagnóstico Diferencial) |
Parte A: Paciente masculino de 4 meses de edad
asiste a consulta externa de pediatría con padre y madre, durante el
interrogatorio los padres indican que lo notaron blandito desde que nació.
No sostiene la cabeza, tiene debilidad. Tarda hasta 45 minutos en alimentarse
de leche materna. Se cansa durante la alimentación y jugando. Parte B: Examen físico: Un adecuado peso y
talla para edad y sexo. Está alerta, consciente y con buen monitoreo visual.
Hace sonidos guturales. Presenta postura en libro abierto. Tiene síndrome
hipotónico. Reflejos miotáticos abolidos. El niño se fatiga durante la
evaluación física y se pone diaforético. CPK: 50U/L, ALT: 116 U/L, AST: 182
U/L, LDH: 1520 U/L. Electromiografía y conducción nerviosa: Signos de
inestabilidad de membrana potenciales de acción de unidad motora de corta
duración y de baja amplitud en vasto medial del cuádriceps, compatible con
miopatía. Neuroconducciones motoras y sensitivas:
normales. Radiografía de tórax: cardiomegalia. EKG: sugiere cardiopatía
hipertrófica. |
|
AME 2 |
|
|
6 |
Parte A: Paciente de 23 meses masculino quien a
los 7 meses se arrastraba hacia atrás, se bajaba de la cama y se sostenía de
pie y se desplazaba en el caminador. A esa edad se cae de la cama y comienza
a perder la fuerza en forma generalizada, llego un tiempo en que NO movía las
piernas. Adecuado lenguaje y desempeño social. Se está secando de las
piernas, están muy delgadas. Poco eleva los hombros y las caderas existe
mejor movilidad de manos y pies. No hay problemas de esfínteres. Parte B: Examen físico: El con mucha dificultad
sube-eleva los miembro superiores e inferiores, de forma incompleta, le falta
un poco en el control cefálico. Sedente con aumento de la cifosis. Hay
disminución del volumen muscular. Importante hipotonía. No se atora cuando
come. Arreflexia generalizada. Agarres lentos sin dismetría. CPK: normal.
Visión y audición normales. Área cognitiva impresiona como normal. |
|
7 |
Parte A: Paciente de 17 meses femenina quien no
ha logrado ponerse de pie sola. Tiene dificultad para masticar. Adecuado
lenguaje y desempeño social. Parte B: Examen físico: Marcada hipotonía, no
logra sostenerse en cuadrúpedo, sedente con aumento de la cifosis. No hay
disminución del volumen muscular. No se encuentran alteraciones sensitivas.
Reflejos osteotendinosos ausentes. CPK normal. |
|
8 (Diagnóstico diferencial) |
Parte A: Paciente de 23 meses masculino quien a
los 20 meses se arrastraba hacia atrás, se bajaba de la cama y se sostenía de
pie y se desplazaba en el caminador. Los padres observan que ahora presenta
dificultad para ponerse de pie y desplazarse en el caminador. No hay
problemas de esfínteres. Parte B: Examen físico: Reflejos levemente
disminuidos, disminución del volumen muscular proximal, pseudohipertrofia
de pantorrillas. Dificultad para pararse del piso. Marcha anormal. CPK en
10,000 u/dL. |
|
9 |
Parte A: Paciente femenino de 3 años con
diagnóstico genético de AME 2. El diagnóstico fue confirmado a los 2 años.
Asiste en compañía de su hermano de 12 años de edad. La preocupación de los padres es que el
niño mayor pueda tener la misma enfermedad. El examen físico del niño mayor
es normal. La fuerza muscular es 5/5 en la escala RMC en los músculos de las
extremidades. Los reflejos
osteotendinosos son normales. La altura del salto vertical es 0,2m. En la
prueba de 6 minutos caminó 620m. |
|
AME 3 |
|
|
10 |
Parte A: Paciente de 6 años, masculino, traído
a la consulta por sus padres por una alteración de la marcha. El niño camina
en punta de pies. El niño empezó a caminar a los 20 meses. Desde el inicio,
observaron la tendencia a caminar en punta de pies. Con adecuado control de
esfínteres. No tenía antecedentes de problemas perinatales, producto de un
primer embarazo a término sin complicaciones. Parte B: Examen físico: En la marcha se observa
un equino dinámico de los tobillos con levantamiento prematuro de los talones
y pie caído en el balanceo. En el examen de fuerza muscular se encuentra
arreflexia generalizada, en la escala MRC: dorsiflexores
y plantiflexores del tobillo 4/5, flexores de
cadera 3/5, extensores de cadera 5, flexores del codo 5, extensores del codo
3/5. No hay alteración de la sensibilidad. Parte C: CK: 800 u/dl. Neuroconducciones:
neuroconducciones sensitivas y motoras de nervios
mediano y cubital normales. En el nervio tibial derecho la amplitud motora se
encontró en 1,5 mV y en el izquierdo 3 mV. Electromiografía de aguja: unidades motoras con
reclutamiento reducido en los músculos deltoides, tibial anterior. No se
observaron potenciales de fibrilación ni ondas agudas positivas. Se
encontraron algunas fasciculaciones en el músculo gastrocnemio derecho. |
|
11 |
Parte A: Paciente de 12 años, masculino, traído
a la consulta por sus padres porque el niño tiene un bajo rendimiento en
educación física. Le observan dificultad para saltar. El niño empezó a
caminar a los 12 meses. No tenía antecedentes de problemas perinatales,
producto de un segundo embarazo a término sin complicaciones. Parte B: Examen físico: la marcha tiene
características normales. El paciente es capaza de saltar en un pie, el
control muscular selectivo dorsiflexor plentiflexor es normal. El salto vertical es de unos 0,12
m. Se encuentra arreflexia rotuliana. En el examen de fuerza muscular, en la
escala RMC: dorsiflexores y plantiflexores
del tobillo 5/5, flexores de cadera 4/5, extensores de rodilla 4+/5
extensores de cadera 5, flexores del codo 5, extensores del codo 4/5. Parte C: CK: Se recibe el resultado de CK: 110
u/dl. Neuroconducciones: neuroconducciones
sensitivas y motoras de nervios mediano y cubital, peroneo y tibial normales.
Electromiografía de aguja: unidades motoras con reclutamiento reducido en el
músculo iliopsoas derecho. No se observaron signos de inestabilidad de
membrana. Prueba de 6 minutos. Las distancias recorridas cada minuto fueron
las siguientes: minuto1= 85m, minuto2= 80m, minuto3= 81m minuto 4=75m minuto
5=70m minuto 6=66m. |
|
12 |
Parte A: Motivo de consulta: Debilidad en las
piernas. Paciente quien refiere cuadro clínico de larga data de inicio no
claro, pero refiere aproximadamente de inicio hace 30 años, caracterizado por
debilidad muscular en miembros inferiores bilateral, de progresión lenta, que
dificultaba actividades básicas como bajar las escaleras con posterior
visualización de movimientos musculares referidos en el deltoides.
Adicionalmente, refiere sensación de calambres. Antecedente de padres
naturales de Capitanejo sin evidencia de consanguinidad entre los padres y
refiere el paciente que los miembros varones de su familia presentan
debilidad muscular en las piernas. Parte B: Examen físico: Alerta, atento,
orientado en sus 3 esferas, movimientos oculares conservados, pares craneales
conservados, fuerza 4/5 en miembros en las 4 extremidades y en músculos
proximales. Reflejos osteotendinosos: +/++++ predominio en miembros inferiores.
Electromiografía de 4 extremidades: Potenciales de acción de unidad motora
polifásicos, prolongados y de bajo voltaje en ambos muslos en contracciones
voluntarias, así como potenciales de acción de unidad motora de alto voltaje
en todos los músculos restantes de brazos y piernas. Parte C: Examen físico: fasciculaciones
visibles del músculo deltoides izquierdo, después de la provocación por
percusión. El signo de Gowers fue positivo. Función
cognitiva superior normal. Creatinina quinasa (CPK): 289 U/L (rango 22 U/L -
198 U/L). Panel por secuenciación de próxima generación para distrofia
muscular de 102 genes: No reporta variantes de significado incierto,
probablemente patogénicas, ni patogénicas. MLPA SMN1 e SMN2: Cero copias del
exón 7 del gen SMN1 en un estado homocigoto y presencia de más de 5 copias de
SMN2. |
|
13 |
Parte A: Paciente de 4 años, masculino, traído
a la consulta por presentar dificultad para subir escaleras. El niño no logra
saltar ni a pie junto, ni en un pie; no corre bien. Los padres refieren que
gateo a los 11 meses, y camino a los 29 meses. El control de esfínteres es
normal. Desarrollo cognitivo normal. Antecedente de parto a término por
cesárea por oligohidramnios, nació deprimido, estuvo 3 días en la UCI, y
describen hipoxia, al parecer hubo broncoaspiración de meconio. Antecedente
familiar negativo para enfermedad neuromuscular. Parte B: Examen físico: Leve displasia pabellón
auricular derecho, aumento de signos blandos en la marcha en talones,
dificultad en salto a pie junto. Esfera mental normal, hipotonía muscular
moderada generalizada con hiperlaxitud ligamentosa, probable hipersensibilidad
táctil en boca y corporal. ROT 1/4 simétricos. Tiene maniobra de Gowers + constante. Debilidad de predominio proximal
leve, más notoria en MMII. Dificultad para subir escalas. Parte C: CPK 11.000 u/dl. Electromiografía de
aguja: muestra enfermedad de fibra muscular. |
|
Caso
clínico |
Descripción |
|
AME 1 |
|
|
1 |
Parte A: Paciente de 6 meses femenina es traída
a consulta por sus padres porque aún no sostiene bien la cabeza, esta
acostada y poco se mueve. La mamá la siente como una gelatina. Sin embargo,
tiene adecuada interacción con el medio. Presenta problemas de succión y en
la consulta notan llanto débil. Antecedente familiar: retardo mental tío
abuelo. Parte B: Examen físico: Ella no sube-eleva los
miembros inferiores, no hay adecuado control cefálico. No hay disminución del
volumen muscular. Se atora cuando come, Dice ma-ma,
pa-pa. Sensibilidad normal. No hay deformidades. Se
observa arreflexia. Ecografía cerebral normal, y audición normal. CPK:
normal. |
|
AME 2 |
|
|
2 |
Parte A: Paciente de 17 meses femenina quien no
ha logrado ponerse de pie sola. Tiene dificultad para masticar. Adecuado
lenguaje y desempeño social. Parte B: Examen físico: Marcada hipotonía, no
logra sostenerse en cuadrúpedo, sedente con aumento de la cifosis. No hay
disminución del volumen muscular. No se encuentran alteraciones sensitivas. Reflejos
osteotendinosos ausentes. CPK normal. |
|
AME 3 |
|
|
3 |
Parte A: Paciente de 12 años, masculino, traído
a la consulta por sus padres porque el niño tiene un bajo rendimiento en
educación física. Le observan dificultad para saltar. El niño empezó a
caminar a los 18 meses. No tenía antecedentes de problemas perinatales,
producto de un segundo embarazo a término sin complicaciones. Parte B: Examen físico: la marcha tiene
características normales. El paciente es capaz de saltar en un pie, el
control muscular selectivo dorsiflexor plantiflexor es normal. El salto vertical es de unos 0,12
m. Se encuentra arreflexia rotuliana. En el examen de fuerza muscular, en la
escala MRC: dorsiflexores y plantiflexores
del tobillo 5/5, flexores de cadera 4/5, extensores de rodilla 4+/5
extensores de cadera 5, flexores del codo 5, extensores del codo 4/5 Parte C: Se recibe el resultado de CK: 110 u/dl.
Neuroconducciones: neuroconducciones
sensitivas y motoras de nervios mediano y cubital, peroneo y tibial normales.
Electromiografía de aguja: unidades motoras con reclutamiento reducido en el
músculo iliopsoas derecho. No se observaron signos de inestabilidad de
membrana. Prueba de 6 minutos. Las distancias recorridas cada minuto fueron
las siguientes: minuto 1= 85m, minuto 2= 80m, minuto 3= 81m, minuto 4=75m,
minuto 5=70m, minuto 6=66m. |