Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v56i2.410
Artíuclo de Investigación
Diagnóstico citogenético de AF en una cohorte de pacientes con características clínicas de sospecha de anemia de Fanconi
Cytogenetic diagnosis of FA in a cohort of patients with clinical characteristics of suspected Fanconi anemia
Olga Morenoa, Angela Paredesa, Adriana Rojasa, Iliana de los Reyesb,c, Ernesto Ruedad, Diego Medinae, Jaime Bernala, Juan C. Prietoa, Roser Pujolf,g, Javier Benítezh, Jordi Surrallésf,i, Fernando Suárez-Obandoa,b.
a. Instituto de Genética Humana, Pontificia Universidad Javeriana, Bogotá D.C. Colombia.
b. Hospital Universitario San Ignacio, Bogotá D.C. Colombia.
c. Hospital Militar Central, Bogotá D.C. Colombia.
d. Hospital Universitario de Santander, Bucaramanga, Colombia.
e. Fundación Valle del Lilli, Cali, Colombia.
f. Centro de investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid España.
g. Servicio de Genética e Instituto de Investigaciones Biomédicas del Hospital de Sant Pau, Barcelona España.
h. CNIO: Centro Nacional de Investigaciones Oncológicas, Madrid España.
i. Joint Research Unit UAB-Sant Pau, Biomedical Research Institute, Institut de Recerca Hospital de la Santa Creu i Sant Pau-IIB Sant Pau, Universitat Autónoma de Barcelona, Barcelona, España.
j. Estadística, Magister en epidemiología clínica, Doctora en estadística. Profesora de la Universidad Nacional de Colombia.
Recibido 08 de octubre de 2022 Aceptado 17 de mayo de 2023
Como Citar: Moreno O, Paredes A, Rojas A, De los Reyes I, Rueda E, Medina D, Bernal J, Prieto JC, Pujol R, Benítez J, Surrallés J, Suárez-
Obando F. Diagnóstico citogenético de AF en una cohorte de pacientes con características clínicas de sospecha de anemia de Fanconi.
Pediatr. 2023;56(2):e410.
Autor para correspondencia: Olga María Moreno Niño
Correo electrónico: moreno-o@javeriana.edu.co
Editor adjunto: Alvaro León Jácome Orozco
Resumen
Antecedentes: la Anemia de Fanconi (AF) es una enfermedad heredada, que afecta la reparación del ADN. Clínicamente es heterogénea; mayoritariamente se presentan malformaciones congénitas, aplasia medular temprana y predisposición a cáncer. El defecto genético causa hipersensibilidad a genotóxicos e inestabilidad cromosómica. Esta característica se considera el mejor marcador diagnóstico; sin embargo, llegar a él puede convertirse en un desafío. Objetivo: caracterizar pacientes con AF mediante pruebas citogenéticas en individuos con rasgos clínicos sugestivos de la enfermedad. Métodos: se analizaron 157 individuos con sospecha clínica de AF, 19 con asociación VACTERL, 15 hermanos, y 34 individuos sanos. Se realizó registro de datos clínicos, y prueba citogenética con Diepoxibutano (DEB). Resultados: se identificaron 43 afectados por AF. La relación de índices en células tratadas con DEB del grupo AF vs. No-AF fue significativamente incrementada, 6.7 veces la proporción de células aberrantes, 48 veces el número de roturas por célula, y 6.3 veces el número de roturas por célula aberrante. En AF la edad media de muestreo fue 9.2 años, la proporción de sexos M:F 1.5:1, consanguinidad en 11 casos. Los sistemas hematológico, esquelético, tegumentario, y urinario estuvieron significativamente alterados. Conclusiones: La AF se identificó en 26 % del grupo de sospecha y en 13 % de hermanos sin sospecha previa. La enfermedad hematológica fue el síntoma más recurrente presente en 93 % de los casos, y fue principalmente la primera sospecha de AF y motivo de estudio genético.
Palabras clave: Anemia de Fanconi, inestabilidad cromosómica, fragilidad cromosómica, asociación VACTERL.
Abstract
Background: Fanconi Anemia (FA) is an inherited disease that affects DNA repair. Clinically, it is heterogeneous; congenital malformations, early bone marrow aplasia, and a predisposition to cancer are present. The genetic defect causes hypersensitivity to genotoxic agents and chromosomal instability; this feature is considered the best diagnostic marker; however, reaching it can be challenging. Objective: To characterize patients with FA using cytogenetic tests in individuals with clinical features suggestive of the disease. Methods: 157 individuals with clinical suspicion of FA, 19 with VACTERL association, 15 siblings, and 34 healthy individuals were analyzed. Recording of clinical data and cytogenetic tests with Diepoxybutane (DEB) were performed. Results: 43 people affected by AF were identified. The ratio of indices in DEB-treated cells of the AF group vs. No-AF was significantly increased: 6.7-fold the proportion of aberrant cells, 48-fold the number of breaks per cell, and 6.3-fold the number of breaks per aberrant cell. In FA, the mean sampling age was 9.2 years, the sex ratio M:F was 1.5:1, and consanguinity was present in 11 cases. The hematologic, skeletal, integumentary, and urinary systems were significantly altered. Conclusions: FA was identified in 26% of the suspected group and in 13% of siblings without prior suspicion. The hematological disease was the most recurrent symptom present in 93% of cases and was mainly the first suspicion of FA and the reason for genetic study.
Key words: Fanconi Anemia, chromosome instability, chromosome fragility, VACTERL association.
Introducción
La Anemia de Fanconi (AF) (OMIM: 227650) es una enfermedad genética de inestabilidad cromosómica, que compromete diversas proteínas involucradas en la reparación del DNA y la regulación del ciclo celular (1-3). Tiene una prevalencia de 1 a 5 afectados por millón y una frecuencia estimada de portadores de 1 en 181 en la mayoría de las poblaciones (4). En algunos grupos étnicos se ha reportado mayor prevalencia por un efecto fundador, aislamiento geográfico, o endogamia (5-7).
Las manifestaciones clínicas son altamente variables y pueden presentarse desde muy severas, con múltiples malformaciones, hasta pacientes con fenotipos normales, con síntomas hematológicos muy tempranos durante la niñez. (8-11). Una característica que contribuye a la heterogeneidad y dificulta el diagnóstico de AF es el solapamiento del fenotipo clínico con síndromes de fallo medular heredados (11), y la asociación VACTERL (12), entre otros síndromes poco comunes (8).
Los afectados por AF presentan hipersensibilidad exacerbada a agentes alquilantes y quimioterapéuticos, que causan daño en el ADN por enlazamiento covalente de la doble hebra o ICL (del inglés: interstrand crosslink); en AF no puede eliminarse el ICL, provocando rotura de las hebras de ADN, reparación no homóloga de extremos rotos, y elevada inestabilidad cromosómica (13-15). En células normales, la reparación de ICLs se activa en fase S y es realizada por la vía de reparación FA o FA/BRCA (FA: del inglés Fanconi Anemia y BRCA: del inglés Breast Cancer, por la participación de BRCA1 y 2) (16,17). Esta ruta en AF está alterada por mutaciones patogénicas en cualquiera de los 22 genes relacionados (FANCA hasta FANCW) cuyas proteínas participan en la vía FA/BRCA (17-19). Las variantes patogénicas en 20 genes (FANC-A,-C,-D1,-D2-E,-F,-G,- I,-J,-L,-M,-N,-O,-P,-Q,-S,-T,-U,-V,-W) son de carácter autosómico recesivo (20); en FANCB recesivo ligado a X (21), y en FANCR autosómico dominante y adquiridas de novo (22,23).
Las nuevas metodologías moleculares para el análisis de variantes patogénicas han contribuido a mejorar el diagnóstico de AF, sin embargo, la prueba citogenética de fragilidad cromosómica por exposición in vitro a agentes inductores de ICL se considera la prueba de oro para evidenciar el fenotipo de inestabilidad (8,19,24). Esta prueba es altamente sensible y puede aplicarse en el diagnóstico prenatal de parejas portadoras de variantes patogénicas, durante el periodo posnatal en individuos con fenotipos clínicos sospechosos y en adultos con infertilidad o reacción adversa a quimio- o radioterapéuticos (8,25,26).
A continuación, se presentan los resultados del primer estudio de una cohorte colombiana de pacientes con diagnóstico o sospecha clínica de AF, y el establecimiento de su fenotipo celular de inestabilidad cromosómica como prueba confirmatoria.
Métodos
Muestra
Estudio prospectivo en pacientes con sospecha de AF, candidatos a prueba citogenética para su diagnóstico. Los pacientes fueron remitidos de la consulta de genética, pediatría, hematología u oncohematología de varias instituciones de salud de Bogotá, D.C. y otras ciudades del país entre 2012 y 2018.
Se establecieron cuatro grupos de estudio: el grupo 1 (G1 o SIAF: Sospecha Inicial de AF) que incluyó 157 casos con diagnóstico clínico o sospecha de AF por características como malformaciones congénitas asociadas a AF o fallo medular no explicado por otras causas; el grupo 2 (G2): 19 casos con asociación VACTERL; el grupo 3 (G3): 15 hermanos de pacientes con AF, y el grupo 4 (G4): 34 individuos sanos (grupo control).
Procedimientos
Las pruebas citogenéticas fueron realizadas en muestras de sangre periférica heparinizada por exposición in vitro a diepoxibutano (DEB) (25). En cada caso se establecieron 4 cultivos de sangre total en medio RPMI 1640 (SIGMA), suero bovino fetal (SFB) (Eurobio) al 10 % y fitohemaglutinina (Gibco), e incubados a 37ºC, CO2 al 5% y humedad de 85 %. 24 horas después, dos réplicas fueron tratadas con DEB (ALFA AESAR) [0.1ug/ml]final y las dos restantes con el diluyente (RPMI) para cultivos no tratados (No-DEB), e incubadas por 48 horas. Simultáneamente, cada prueba fue acompañada por cultivos adicionales de una línea celular linfocitaria AF como testigo positivo y control de actividad del clastógeno, y de sangre de un individuo sano, sometidas al mismo protocolo. Las células en metafase fueron obtenidas por tratamiento con colchicina (SIGMA) [0.01ug/ml] final, solución hipotónica (KCl (MERCK) 0.075M) y fijación con metanol:ácido acético (MERCK) 3:1. Los extendidos cromosómicos fueron teñidos con Giemsa al 4 % (MERK) (25).
En cada caso y por tratamiento se evaluó la inestabilidad cromosómica en 50 o 100 metafases (Figura 1). La hipersensibilidad al clastógeno se estableció mediante el cálculo de los índices: 1) porcentaje de células aberrantes (células con al menos uno de los siguientes eventos: rotura cromatídica o isocromatídica, fragmento, figura radial, deleción, translocación, anillos, dicéntricos; los gaps cromatídicos o isocromatídicos con espesor menor al grosor de la cromátide no fueron considerados); 2) roturas por célula (total de roturas cromosómicas obtenidas en relación con el total de células evaluadas); 3) roturas por célula aberrante (total de roturas obtenidas en relación con el total de células aberrantes); 4) porcentaje de células multiaberrantes (células con dos o más roturas), y 5) índice de fragilidad cromosómica (IFC) (% de células aberrantes x número de roturas por célula multiaberrante) (27). Adicionalmente se hizo bandeo G (BG) con HCl (MERCK) 0.025N, y coloración con Wright (MERCK), y se describió el cariotipo según International System for Human Cytogenomic Nomenclature 2020 (ISCN 2020). Cuando la prueba citogenética fue no concluyente, se analizaron fibroblastos para descartar mosaicismo.
Análisis estadísticos
Los datos de roturas cromosómicas fueron descritos como frecuencias absolutas y relativas. Se aplicó la prueba de Wilcoxon para muestras independientes con una significancia del 5 %. Los datos clínicos fueron tabulados y analizados por estadísticas descriptivas y prueba exacta de Fisher. Los análisis fueron realizados en el paquete estadístico R, versión 3.5.1.
Aspectos éticos
Todos los participantes y sus tutores recibieron el proceso de consentimiento informado. Los datos clínicos fueron obtenidos de la historia clínica remitida con la muestra, o de la valoración del paciente por un genetista clínico del equipo de investigación. El estudio fue aprobado por el comité de ética de la Facultad de Medicina de la Pontificia Universidad Javeriana de Bogotá, D.C. y el Hospital Universitario San Ignacio (HUSI), CIE-2011/29, -2014-107 y -2014/150.
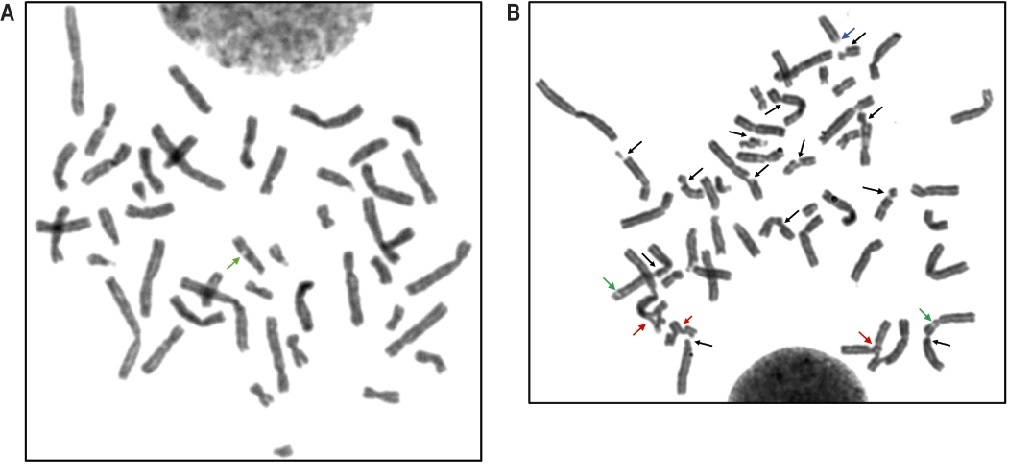
Figura 1. Inestabilidad cromosómica en anemia de Fanconi.
Fotografías de metafases de paciente con AF de cultivos no tratados y tratados con DEB A. Metafase de células no tratadas con presencia de gap cromatídico (flecha verde); B. Metafase con roturas cromosómicas y figuras radiales de cultivo tratado del mismo paciente. Roturas cromatídicas (flechas negras), figuras radiales (flechas rojas), fragmento acéntrico (flecha rosada). Los gaps (flechas verdes) no son contabilizados como evento de rotura para AF. Coloración homogénea con Giemsa, microscopio ZEIZ Axioplan2, objetivo 100x.
Resultados
Características de los sujetos analizados
Las características demográficas son descritas en la tabla 1. Los 157 casos de G1 pertenecieron a 152 familias. La consanguinidad parental fue confirmada en 9 (7.3 %) familias de 124 en las que se obtuvo información.
La característica clínica más común en G1 fue la alteración del sistema hematopoyético en 125 casos (79.6 %), con edad promedio de inicio de 6.5 años, seguido por la afectación de los sistemas esquelético en 68 sujetos (43.3 %), tegumentario en 55 casos (35 %), talla baja en 49 afectados (31.2 %), y alteraciones faciales en 41 pacientes (26.1 %). En G2 ningún caso tuvo anomalía sanguínea o historia de consanguinidad, el número de alteraciones congénitas asociadas a la asociación VACTERL se presentó desde 3 hasta 6, y en 9 casos afectó miembros superiores, característica fuertemente asociada a AF. Otras características clínicas y genéticas de G2 fueron publicadas en Moreno et al. (28). En G3 fueron hermanos de 10 casos AF confirmados en este estudio (10 familias, 6 consanguíneas) sin sospecha de AF.
Análisis citogenético
Los índices calculados son presentados en la tabla 2. Los valores de proporción de células aberrantes (%), roturas por célula, y roturas por célula aberrante en cultivos tratados en comparación con los no tratados con DEB (Ver figura 2), fueron significativamente incrementados en 43 casos pertenecientes a 40 familias y permitieron clasificarlos como afectados por AF: 41/157 (26.1 %) de G1 y 2/15 (13 %) de G3; 13 casos de G1 tuvieron un resultado no concluyente.
Los individuos restantes de G1, 103/157 (65.6 %), los 19 casos de asociación VACTERL, (100 %), los sujetos de G3, 13/15 (87 %), y los 34 de G4 (100 %), fueron resistentes al genotóxico, con valores en los índices evaluados muy similares y diferencias no significativas (p>0.05) entre ellos; así, fueron agrupados como no afectados por AF (no-AF).
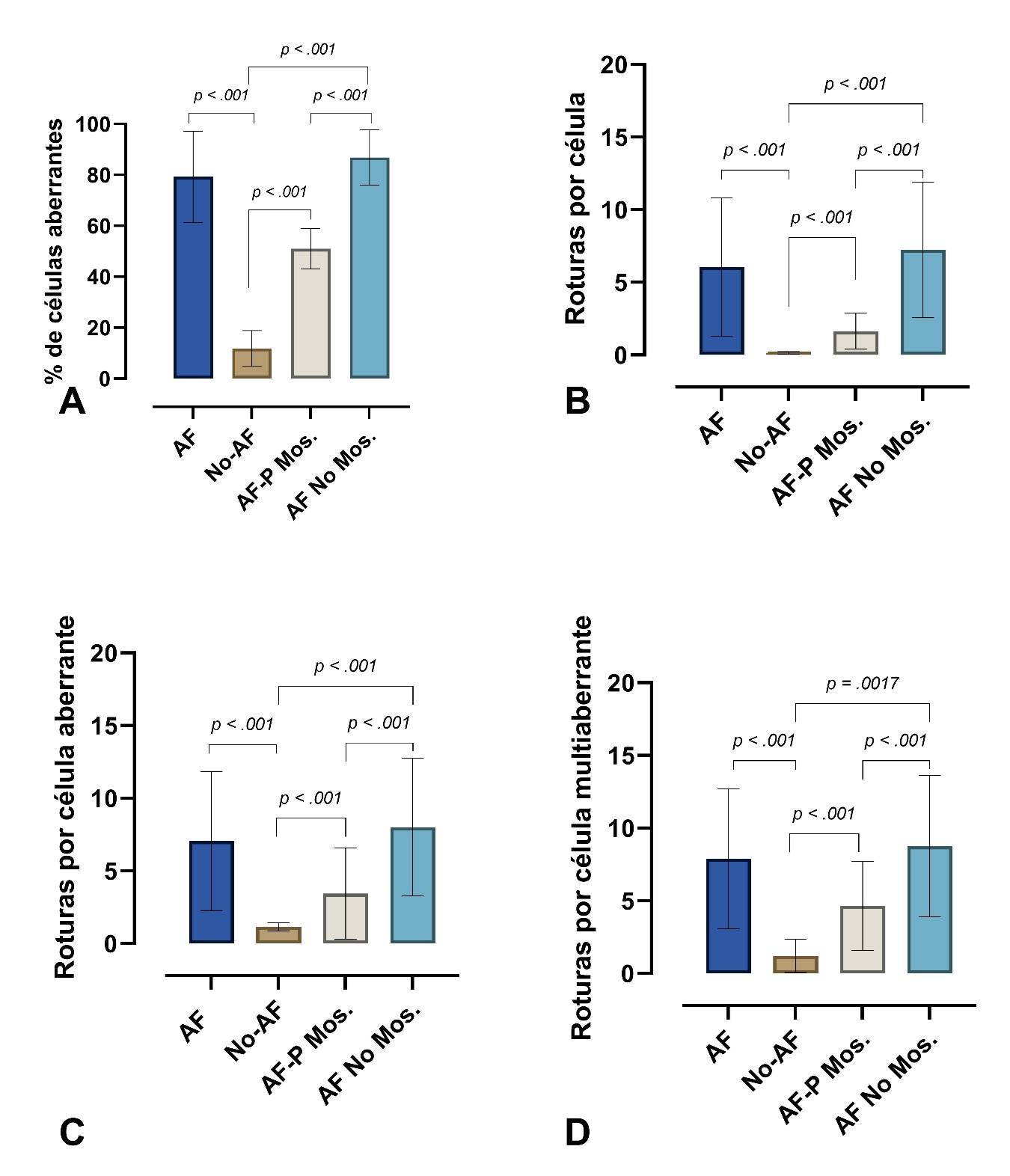
En cultivos tratados, los valores medios de roturas en el grupo AF fueron significativamente incrementados en relación con los del grupo no-AF. El incremento medio de porcentaje de células aberrantes fue de 7 veces (medias: 79.3 % y 11.83 % respectivamente.), roturas por célula de 48 veces (medias: 6.06 y 0.14 respectivamente), roturas por célula aberrante de 6.3 veces (medias: 7.05 y 1.16 respectivamente), e IFC de 22 veces (medias: 663.74 y 30.18 respectivamente).
Las figuras radiales cuya presencia y complejidad son un distintivo de la AF se presentaron en células tratadas con DEB desde 5 hasta 240 por caso (media por célula: 0.72), mientras que el grupo no-AF mostró de 0 a 1 (media por célula: 0.0027). El mosaicismo somático en células sanguíneas de AF se consideró cuando el porcentaje de células aberrantes fue menor de 60 % (27); 9 casos con índices de células aberrantes en un rango entre 39 % y 60% fueron clasificados como posibles mosaicos (P-mos); 34 casos claramente fueron no-mosaicos (No-mos), con índices de células aberrantes superiores a 60% (rango 61 % a 100 %). Los valores medios de roturas por célula y de roturas por célula aberrante de P-mos con respecto a AF No-mos fueron significativamente disminuidos (p < 0.005), y significativamente diferentes de los del grupo No-AF (p < 0.005) (Ver tabla 2 y figura 3).
Trece casos fueron clasificados inicialmente como inconclusos. Estos casos tuvieron un conteo aumentado de roturas cromosómicas tanto espontáneas como inducidas por DEB, sin embargo, las características de las aberraciones no fueron clásicas de AF. Posteriormente, en cuatro casos se descartó AF; en los otros nueve no fue posible confirmar el resultado.
La presencia de inestabilidad espontánea en células no tratadas fue altamente heterogénea, con valores para células aberrantes desde 0% hasta 61% para casos de AF (Ver tabla 2, figura 2). Estos valores presentaron solapamiento con los de No-AF en todos los índices, y no hubo una separación clara entre afectado y no afectado a pesar de las diferencias significativas (p < 0.005) en los valores medios entre los dos grupos. El análisis de cariotipo detectó anomalías cromosómicas en 5 sujetos (4 de G1 y 1 de G4) y variantes en heterocromatina constitutiva en 3 participantes.
Tres casos fueron AF, uno presentó un clon hiperdiploide:5 4~56,XY,+3,+7,+8,+10,+13,+16,+22,+mar1,+mar2[Cp6]/46,XY[94] en sangre, y los dos restantes variantes de la heterocromatina 46,XY,inv(9)(p12q13) y 46,XY Yqspat, consideradas no patogénicas.
Características demográficas y clínicas del grupo de casos AF
En la tabla 1 se describen las características demográficas para AF. La edad media a la toma de la muestra del grupo AF fue de 9.2 años (mediana: 8). El número de casos de sexo masculino con relación a los de sexo femenino fue significativamente diferente con una proporción 1.5:1 (p < 0.05) y opuesta a la del grupo SIAF No-AF. Consanguinidad parental fue confirmada en 11/43 (25.6 %) casos de 8 familias, 23/43 (53.5 %) informaron no consanguinidad, y 9/43 (21 %) no informaron.
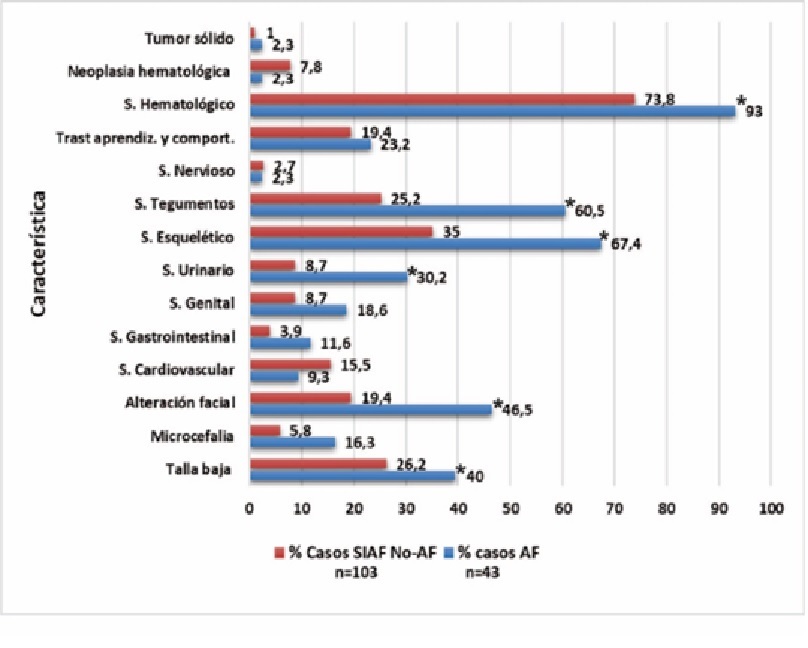
En AF prácticamente todos los sistemas fueron afectados, sin embargo, fue mayor en los sistemas (de mayor a menor) hematológico (p < 0.05), esquelético (p < 0.005), tegumentario (p < 0.005), fenotipo facial (p < 0.005), la talla (p < 0.05) y urinario (p < 0.005) (ver figura 4). El sistema hematológico presentó afectación en 40 casos (93 %), mayoritariamente por fallo de la médula ósea (MO) y trombocitopenia. Neoplasias fueron informadas en un caso con leucemia linfocítica aguda (LLA) y otro con osteosarcoma.
Tabla 1. Características demográficas de la muestra analizada pre- y posanálisis citogenético.
|
Característica |
Grupos preanálisis |
|
Grupos posanálisis |
|||||||
|
G1 (SIAF) |
G2 |
G3 |
G4 |
SIAF No- AF |
AF |
|
||||
|
AF |
No-Mosaico |
P- Mosaico |
||||||||
|
No. de casos
|
157 |
19 |
15 |
34 |
103 |
43 |
34 |
9 |
||
|
Edad al examen (Media/rango) |
10.5 (0.08-67) |
2,8 (<1-17) |
13 (<1-8) |
20 (4-47) |
10.51 (<1-58) |
9.2 (<1 -7) |
10.4 (<1-57) |
5.6 (<1-21) |
||
|
Sexo M/F proporción
|
73/84 1:1.2 |
9/10 1:1.1 |
8/7 1.1:1 |
21/13 1.6:1 |
42/61 1:1.5 |
26/17 1.5:1 |
20/14 1.4:1 |
6/3 2:1 |
||
|
Edad inicio enfermedad Hematológica |
6.85 (<1-57) |
N.A. |
N.A. |
N.A. |
6,5 (<1-56) |
6.9 (<1-54) |
7.41 (2-54) |
5.41 (<1-17) |
||
|
Consanguinidad parental informada Si/No |
13/124 |
0 |
6/15 |
0 |
2/78 |
11/23 |
9/16 |
2/7 |
||
G1: Grupo de casos con diagnóstico clínico o sospecha de AF; G2: Grupo de casos con asociación VACTERL, G3: Grupo de hermanos de casos AF; G4: Grupo sano. SIAF: sospecha inicial de anemia de Fanconi; AF: anemia de Fanconi; P-Mosaico: probable mosaico; M/F: masculino/femenino; N.A.: No aplica.
Tabla 2. Resultados completos del estudio citogenético en todos los grupos evaluados con prueba de fragilidad.
|
|
% Células aberrantes |
Roturas por célula |
Roturas/célula aberrante |
% Células multiaberrantes |
Roturas/célula multiaberrante |
IFC |
||||||||||||
|
Grupo |
n |
Media |
Intervalo |
n |
Media |
Intervalo |
n |
Media |
Intervalo |
n |
Media |
Intervalo |
n |
Media |
Intervalo |
n |
Media |
Intervalo |
|
Tratados con DEB |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1. No-AF (total) |
169 |
11.83 |
2 - 35 |
169 |
0.14 |
0.016 – 0.52 |
169 |
1.16 |
1 – 3.5 |
169 |
1.83 |
0 - 14 |
93 |
2.19 |
2 - 5 |
73 |
28.37 |
4 – 74.5 |
|
1.2 Grupo SIAF No- AF |
103 |
11.99 |
2 - 35 |
103 |
0.14 |
0.016 – 0.45 |
103 |
1.17 |
1 – 3.5 |
103 |
1.91 |
0 - 11 |
57 |
2.21 |
2 - 4 |
60 |
30.80 |
4 – 74.5 |
|
1.1 Hermanos |
13 |
13 |
4 - 26 |
13 |
0.14 |
0.05 – 0.28 |
13 |
1.07 |
1 – 1.33 |
13 |
1.08 |
0 - 5 |
6 |
2.03 |
2 – 2.2 |
6 |
25.27 |
8 – 39.6 |
|
1.3 VACTERL |
19 |
10.37 |
2 - 26 |
19 |
0.12 |
0.02 – 0.37 |
19 |
1.14 |
1 – 1.5 |
19 |
1.32 |
0 - 9 |
9 |
2.40 |
2 – 5 |
10 |
26.94 |
4 – 54.89 |
|
1.4 Grupo sano |
34 |
11.71 |
2 - 26 |
34 |
0.14 |
0.02 – 0.52 |
34 |
1.19 |
1 – 2 |
34 |
2.15 |
0 - 14 |
21 |
2.11 |
2 – 3 |
20 |
31.44 |
8 – 66.86 |
|
2. AF Total |
43 |
79.30 |
39 - 100 |
43 |
6.06 |
0.69 – 22.01 |
43 |
7.05 |
1,64 – 22.23 |
43 |
66.38 |
13 - 100 |
43 |
7.89 |
2.84 - 23 |
43 |
663.74 |
131.15 - 2201 |
|
2.1 AF No-Mosaico |
34 |
86.79 |
67 - 100 |
34 |
7.22 |
1.72 – 22.01 |
34 |
8.01 |
2,49- 22.23 |
34 |
75.39 |
42 - 100 |
34 |
8.76 |
3.32 - 23 |
34 |
778.97 |
232.4 - 2201 |
|
2.2 AF P-Mosaico |
9 |
51 |
39 - 60 |
9 |
1.66 |
0.69 – 4.74 |
9 |
3.42 |
1,64 – 11.56 |
9 |
32.33 |
13 - 60 |
9 |
4.63 |
2.84 – 12.39 |
9 |
228.42 |
131.15 - 508 |
|
Espontáneos – No tratados con DEB |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1. No-AF (total) |
169 |
4.37 |
0 - 22 |
169 |
0.05 |
0 – 0.24 |
135 |
1.07 |
1 - 2 |
169 |
0.27 |
0 - 4 |
29 |
2.14 |
2 - 4 |
29 |
16.29 |
2 - 44 |
|
1.2 Grupo SIAF No- AF |
103 |
4.67 |
0 - 18 |
103 |
0.05 |
0 – 0.19 |
85 |
1.07 |
1 - 2 |
103 |
0.27 |
0 - 3 |
20 |
2.20 |
2 - 4 |
19 |
16.32 |
4 - 36 |
|
1.1 Hermanos |
13 |
4.62 |
0 - 11 |
13 |
0.05 |
0 – 0.12 |
11 |
1.05 |
1 - 1,5 |
13 |
0.23 |
0 - 2 |
2 |
2 |
2 - 2 |
2 |
15 |
8 - 22 |
|
1.3 VACTER |
19 |
2.05 |
0 - 10 |
19 |
0.02 |
0 – 0.1 |
13 |
1.17 |
1 - 2 |
19 |
0.32 |
0 - 4 |
3 |
2 |
2 - 2 |
3 |
4 |
2 - 6 |
|
1.4 Grupo sano |
34 |
4.65 |
0 - 22 |
34 |
0.05 |
0 – 0.24 |
26 |
1.01 |
1 - 1,17 |
34 |
0.24 |
0 - 2 |
4 |
2 |
2 - 2 |
4 |
26 |
10 - 44 |
|
2. AF Total |
43 |
18.59 |
0 - 61 |
43 |
0.26 |
0 – 1.32 |
42 |
1.28 |
1 - 2,75 |
43 |
4.63 |
0 - 34 |
32 |
2.27 |
2 - 4,5 |
32 |
51.5 |
10 – 189.71 |
|
2.1 AF No- Mosaico |
34 |
21.37 |
4 - 61 |
34 |
0.30 |
0.04 – 1.32 |
34 |
1.26 |
1 - 2,16 |
34 |
5.44 |
0 - 34 |
25 |
2.24 |
2 - 3,11 |
25 |
60.24 |
10 – 189.71 |
|
2.2 AF P-Mosaico |
9 |
8.11 |
0 - 13 |
9 |
0.11 |
0 – 0.22 |
8 |
1.36 |
1 - 2,75 |
9 |
1.56 |
0 - 4 |
7 |
2.36 |
2 - 4,5 |
7 |
20.29 |
12 - 36 |
DEB: diepoxibutano; IFC: Índice de fragilidad cromosómica; n: Número de individuos; SIAF: sospecha inicial de anemia de Fanconi.
Figura 2. Análisis de roturas cromosómicas.
Resultados de los análisis de roturas cromosómicas e índices calculados en cultivos no tratados (No-DEB) en azul, y tratados (DEB) en rojo en cada uno de los 43 casos AF. IFC: Índice de Fragilidad Cromosómica.
Figura 3. Medias de los valores de los diferentes índices evaluados en células expuestas a los tratamientos.
Diagrama de cajas que representa las medias de los valores de los diferentes índices evaluados en células expuestas a DEB en los grupos AF (total), No-AF, AF No-Mosaico, y AF P-mosaico (probable mosaico). En todos los grupos las diferencias entre ellos fueron altamente significativas P < 0.001. A. % de células aberrantes; B. Roturas por células; C. Roturas por célula aberrante; D. Roturas por célula multiaberrante.
Figura 4. Características clínicas de los dos grupos AF y SIAF No-AF.
La figura muestra en porcentajes las frecuencias de casos con cada característica y en cada grupo. Las diferencias significativas entre los dos grupos se muestran con la estrella. SIAF No-AF: Sospecha inicial de AF no anemia de Fanconi después de los análisis.
Discusión
El fenotipo de inestabilidad genómica en los pacientes con AF puede ser determinado en cromosomas metafásicos aplicando la metodología citogenética apropiada (29). Aunque los pacientes con AF pueden expresar espontáneamente elevada tasa de roturas (30), no siempre ocurre, como se ha confirmado en este estudio, en donde las tasas de rotura variaron ampliamente. Por esta razón, los protocolos estandarizados con agentes inductores de ICLs permiten identificar el fenotipo de inestabilidad, expresado preferencialmente en roturas cromatídicas y configuraciones radiales (19,29,31). A pesar de estos protocolos, se estima que existe un subdiagnóstico de la enfermedad (19,32,33).
La confirmación de AF es de extrema importancia para el manejo y seguimiento del afectado, debido al alto riesgo de muerte por fallo medular severo, predisposición a cáncer, y por las implicaciones que tiene a nivel familiar (8). Por esta razón, y la necesidad de conocimiento, se presenta el análisis citogenético de 157 pacientes reclutados por diagnóstico o sospecha clínica de AF, y de 19 casos con asociación VACTERL, con confirmación de AF en 41 casos (26 %), y dos hermanos de afectados, previamente no sospechosos.
Los resultados reportados aquí son similares a los publicados por Esmer et al. (34)hyperpigmentation, small stature, congenital abnormalities, and pre-disposition to neoplasia characterize Fanconi anemia (FA quienes identificaron 27.8 % (15/54) de afectados por AF en pacientes mexicanos con aplasia medular y alta sospecha clínica de AF. A diferencia del presente estudio, estos investigadores hallaron también un afectado por AF en su grupo de casos con asociación VACTERL; esto ratifica la necesidad de la prueba para AF durante el diagnóstico diferencial de VACTERL (12,34,35)245 cases of FA reported in the literature from 1927 to 2012 identified 108 cases with at least 3 of the defining features of VATER association; only 29 had been so noted by the authors. The FA VATER signature was the significantly higher frequency of renal and limb (radial and/or thumb.
Es infrecuente que dentro del diagnóstico diferencial de la asociación VACTERL se incluya la prueba para AF, sin embargo, el alto riesgo de enfermedad hematológica y de cáncer en los afectados por AF justifica su inclusión. Por otra parte, el hallazgo de AF en hermanos, muestra la importancia del asesoramiento y seguimiento a la familia del paciente AF para la detección oportuna de afectados no sospechosos. En este contexto, un caso de esta cohorte producto de una pareja consanguínea en primer grado, y producto de un embarazo gemelar cuyo hermano gemelo murió por múltiples malformaciones días después del nacimiento, la sospecha de AF en el sobreviviente solo se dio hasta la aparición del fallo medular a los 6 años; paradójicamente, luego de la confirmación con prueba citogenética del gemelo sobreviviente, la prueba en su hermano mayor, de 8 años, no sospechoso, resultó también positiva. Por otro lado, dado que los hermanos son los primeros candidatos como donantes de células madre hematopoyéticas para el tratamiento del fallo medular, es importante establecer su estado de afectado o sano (36). AF es mayoritariamente autosómica recesiva, por lo tanto, cada hermano de un afectado tendrá un riesgo de 25 % de AF que debe ser determinado (1).
En grupos de pacientes con características similares a las presentadas aquí, Auerbach et al. (25) con pacientes remitidos a IFAR (del inglés: International Fanconi Anemia Registry), Korgaonkar et al. (37)9 (4% en pacientes del Departamento de Hematología del Hospital K.E.M en Mumbai-India, y Cirkovic et al. (38) en pacientes Serbios, identificaron 61.6% (202/328), 34.4 % (67/195) y 15.2 % (10/66) de casos AF respectivamente. Es de resaltar que los resultados reportados por estos grupos, junto con los informados en México y los presentados aquí varían ampliamente, probablemente debido a la sutileza del análisis clínico diferencial previo a la confirmación citogenética. Particularmente, el número de casos diagnosticados en IFAR difieren en gran medida de los demás, diferencia que podría deberse a las características de los pacientes remitidos a IFAR, como por ejemplo, el descarte previo de otras aplasias medulares heredadas (39).
El presente grupo de pacientes tuvo una sospecha clínica de AF muy heterogénea, desde casos con diagnóstico clínico de AF y prueba citogenética previa (no concluyente en algunos casos), hasta casos con solo aplasia medular que requerían del diagnóstico diferencial. De forma particular, el 21 % contaba con una prueba citogenética previa al ingreso al estudio y el análisis aquí realizado identificó algunas dificultades para el diagnóstico de AF y la posibilidad de un subdiagnóstico (40). En algunos casos se identificó que, a pesar de la alta sospecha clínica, las pruebas citogenéticas confirmatorias fueron fallidas. Las posibles causas identificadas fueron: 1) no especificación clara en la orden médica sobre un estudio de fragilidad para AF, 2) uso del clastógeno no adecuado para la inducción de ICLs o pérdida de actividad de este, y 3) prueba de inestabilidad no adecuada para AF. DEB y MMC son ampliamente usados para el diagnóstico de AF y ambos ofrecen sensibilidad, especificidad y reproducibilidad cuando se aplican los protocolos estandarizados (8). Auerbach (25,41) sin embargo, recomienda precaución al utilizar estos clastógenos especialmente la MMC, debido a que ligeras variaciones en su concentración pueden generar falsos negativos o falsos positivos, mientras que DEB es más estable y reduce esta posibilidad (42,43).
Durante el diagnóstico de AF por pruebas citogenéticas o moleculares pueden presentarse mosaicismos somáticos en células sanguíneas de los pacientes y dificultar el diagnóstico (42). Esta condición también denominada «terapia génica natural », surge en una célula madre del tejido hematopoyético por mutaciones que corrigen el defecto genético en uno de los alelos alterados y restablece la función reparadora en la célula y su descendencia por expansión clonal (43–45)bone marrow failure (BMF. En AF el fenotipo celular en mosaico es evidenciado por la presencia de poblaciones celulares sanguíneas genéticamente distintas y una alta variación en la sensibilidad a los agentes inductores de ICLs (27,42,46). Esta variación depende del tamaño del clon corregido y puede alcanzar desde un 40% hasta el 100 % de células insensibles al clastógeno (47)with nonhematologic symptoms of Fanconi anemia (FA.
El mosaicismo en AF puede presentarse hasta en un 2 5% de los casos, por lo cual cuando los resultados de las pruebas diagnósticas no son los clásicos o son no concluyentes en un sospechoso, debe considerarse esta posibilidad (46). En este estudio, 9/43 casos (20.9%) fueron clasificados como P-mos, concordando con los reportes en otras poblaciones (27,46). Este grupo registró entre un 40 % y 61 % de células insensibles al clastógeno ( < 60% de células aberrantes) y la línea celular aberrante mantuvo una sensibilidad al daño relativamente alta, aunque con una disminución significativa en los valores medios con relación al grupo AF No-mos. En AF el fenotipo en mosaico se ha asociado con aparición tardía de síntomas hematológicas, protección contra complicaciones severas, y reducción en la necesidad de trasplante (48)bone marrow failure (BMF; en contraste con lo anterior, nuestro grupo P-mos mostró un promedio de edad de inicio menor de la del grupo No-mosaicos, por lo tanto, se sugiere evaluar las causas de inicio más temprano de síntomas y realizar seguimiento hematológico continuo.
Este resultado concuerda con el grupo clasificado por Castella et al. (27) como probable mosaico (40 % a 60 %), y no se registró un grupo separado definido como mosaicos según el criterio de estos investigadores, en dónde el rango de células aberrantes fue reportado entre el 10 % y el 38 % (27). De otra parte, un grupo de 13/157 (8.3 %) casos fue clasificado como “inconcluso” con resultados de índices de % de células aberrantes entre 20 % y 46 %. Estos resultados claramente se solaparon con los resultados del grupo No-AF y P-Mos, sin embargo, debido a la presencia mayoritaria de aberraciones no típicas de anemia de Fanconi como fragmentos acéntricos, reordenamientos cromosómicos estructurales y ausencia de figuras radiales se clasificó como inconcluso.
De manera particular, un caso, en ambos tipos de cultivo mostró elevada tasa de daño típico de exposición a radiación y prueba negativa en fibroblastos, probablemente por efecto del tratamiento con quimioterapéuticos para un meduloblastoma. Otros tres casos, uno con 35 %, y dos, cada uno con 46 % de células aberrantes, se les confirmó síndrome de Emberger (49), síndrome de Wiskott Aldrich (50) y síndrome de desnutrición severa, respectivamente. Estas observaciones hacen considerar que en algunos casos otras condiciones genéticas, fisiológicas, de fallo medular, o del ambiente en los pacientes, distintas a AF, pudieran generar una moderada respuesta reparadora del daño, dificultando su clasificación por prueba citogenética.
Se ha informado que, en aproximadamente un 10 % de los pacientes con síndromes de fallo medular heredado (no-AF), o en heterocigotos para las mutaciones AF puede hallarse un ligero aumento en la frecuencia de células aberrantes y de roturas, que pueden dificultar el diagnóstico (42). En este contexto, es importante la experticia en el reconocimiento de los eventos citogenéticos de inestabilidad en AF durante los análisis, realizar prueba en fibroblastos, o aplicar metodologías moleculares como whole exome sequence (WES) para la definición diagnóstica cuando los resultados son no concluyentes (24,29,42).
Castella et al. (27) han propuesto el uso del IFC para clasificar a los pacientes como AF durante el análisis de la prueba citogenética; así, en pacientes AF españoles confirmados con prueba molecular establecieron valores de IFC < 40 para individuos no-AF y >55 para individuos AF incluidos los mosaicos. En el presente estudio, el IFC en individuos sanos fue muy variable y más alto del propuesto (rango: 0 a 66.86). Claramente los valores de IFC en individuos AF y AF P-mos (rango: 131.2 a 2201) los diferenciaron de los individuos No-AF, sin embargo, no se estableció un valor umbral para separar la población No- AF de AF debido a la no identificación de individuos AF-mosaico con valores de células aberrantes menores de 40 % (27,42,46); de esta manera, en casos con alta sospecha de AF y prueba no concluyente se hace necesario aplicar otras pruebas confirmatorias.
El hallazgo de un clon anormal hiperdiploide en sangre periférica (6 %: 6/100 metafases) de un caso AF, aunque es raro, podría relacionarse con evolución clonal en MO, o progresión neoplásica, dada la elevada predisposición en AF a síndrome mielodisplásico (SMD) y leucemia mieloide aguda (LMA) (51,52) progressive marrow failure and predisposition to myelodysplastic syndrome (MDS. En AF la evolución clonal de alteraciones cromosómicas como ganancia de 1q, 3q y 13q, pérdida de 7, 7q o 20q, pueden indicar progresión maligna, siendo 3q especialmente crítico (51,53,54). Por esta razón, el hallazgo de estas alteraciones en LMA de novo puede ser un indicador de AF que debe ser confirmado (54,55)ataxia telangiectasia (A-T. El caso descrito aquí fue informado con trombocitopenia, y el clon anormal no tiene las características para SMD o LMA, probablemente se trate de un clon transitorio (51), o de un clon que delinee una neoplasia como LLA en la que la hiperdiploidía es común (56). LLA es poco común en AF, pero se ha reportado en algunos casos (52,57)332 developed hematologic abnormalities at a median age of 7 years (range, birth to 31 years, así que en casos como el aquí descrito se recomienda vigilancia de la MO y seguimiento para progresión clonal (51), lo cual no fue posible por pérdida de comunicación con el paciente.
Las manifestaciones fenotípicas del grupo AF involucraron mayoritariamente el sistema esquelético, tegumentario, fenotipo facial, talla, y sistema urinario, en concordancia con lo previamente informado (23,58). El sistema endocrino en particular fue la excepción debido a que no se hallaron datos en las historias clínicas, a pesar de que se ha reportado su afectación hasta en un 81% de los pacientes AF (8,59). En relación con lo anterior, la talla baja, que podría estar asociada a deficiencia de la hormona de crecimiento y disfunción tiroidea, se ha reportado en más del 50 % de los casos, y aunque en este estudio fue menor (40%), podría evidenciar el compromiso del sistema endocrino, lo cual deberá confirmarse con las pruebas apropiadas (59,60).
La anomalía hematológica fue la característica clínica más prevalente, presentándose en el 93 % de los pacientes a un promedio de edad de inicio de los síntomas de 6.9 años ( < 1 a 54), concordante con lo reportado para AF; se ha establecido que los pacientes a una edad promedio de 7 años podrían haber desarrollado algún tipo de anomalía hematológica, cuyo riesgo a los 40 años es del 90 % (52,57)a prospectively collected database of FA patients, allows us the unique opportunity to analyze the natural history of this rare, clinically heterogeneous disorder in a large number of patients. Of the 754 subjects in this study, 601 (80%. Es de resaltar que la edad promedio a la toma de la muestra fue de 9.2 años ( < 1 a 57), lo cual sugiere que en algunos casos la sospecha de AF ocurrió hasta la aparición de anomalía hematológica, y que, además, la confirmación por prueba citogenética fue mucho más tardía.
El retraso en la confirmación diagnóstica de los pacientes es común en otras poblaciones y es más notorio en fenotipos leves (10,32,61). Estas observaciones refuerzan la importancia de implementar protocolos que contemplen el diagnóstico diferencial de AF en diferentes escenarios clínicos. De manera particular los familiares de algunos pacientes concordaron en que los primeros síntomas hematológicos coincidieron con cuadros infecciosos de dengue. Probablemente en regiones endémicas de dengue, la adquisición de la infección por los pacientes en proceso de agotamiento de su MO, previo a un diagnóstico de AF, solape los síntomas del fallo medular (trombocitopenia o leucopenia) con un cuadro sintomático de dengue, o, por el contrario, la infección exacerbe el agotamiento de las células madre y dé paso a una franca expresión de los síntomas hematológicos.
Conclusión
Este es el primer reporte de estudio en AF en población colombiana. La prueba citogenética permitió identificar la población afectada por AF dentro de un amplio grupo de pacientes con características clínicas incluidas dentro del espectro de AF que pueden compartirse con otras entidades clínicas. Se destaca de manera particular la identificación de hermanos afectados a quienes previamente no se les había sospechado AF, resaltando la importancia del árbol genealógico luego de confirmarse un probando. El análisis de cariotipo sumado al de fragilidad cromosómica podría brindar información adicional sobre el estado del paciente, y aunque en este protocolo no se estudió cariotipo en MO de los pacientes AF, este es altamente recomendado para establecer evolución clonal de alteraciones cromosómicas y vigilar tempranamente el riesgo de progresión maligna.
Financiación
Pontificia Universidad Javeriana, ID 00004230 Ref: 2011/29, ID 00006453 Ref: 2014/150 y Pontificia Universidad Javeriana/Hospital Universitario San Ignacio, ID 00006284 Ref: 2014-107.
Agradecimientos
A todos los pacientes y a sus familias por su autorización y participación en el estudio, a las diferentes instituciones de salud y a todos los especialistas clínicos en hematología, oncología, y pediatría que remitieron a los pacientes para el estudio diagnóstico o confirmatorio de AF. Así mismo a los Genetistas y Residentes de la Especialidad de Genética Clínica del Instituto de Genética Humana de Pontificia Universidad Javeriana sede Bogotá, por la valoración de los pacientes y la interpretación de historias clínicas. Al grupo de investigación en enfermedades onco-hematológicas, y síndromes de reparación del DNA y predisposición al cáncer de Institut de Recerca Hospital de la Santa Creu i Sant Pau-IIB Sant Pau, en Barcelona España, por el apoyo y orientación en el desarrollo de la investigación.
Conflictos de interés
Los autores declaran no tener conflictos de interés.
Referencias
1. Fanconi Anemia - GeneReviews® - NCBI Bookshelf [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK1401/ citado 2022 Jul 27].
2. Sumpter R, Levine B. Novel functions of Fanconi anemia proteins in selective autophagy and inflammation. Oncotarget 2016; 7(32):50820–1.
3. Milletti G, Strocchio L, Pagliara D, Girardi K, Carta R, Mastronuzzi A, et al. Canonical and noncanonical roles of fanconi anemia proteins: Implications in cancer predisposition. Cancers (Basel) 2020; 12(9):2684.
4. Rosenberg PS, Tamary H, Alter BP. How High are Carrier Frequencies of Rare Recessive Syndromes? Contemporary Estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A 2012; 155(8):1877–83.
5. Callén E, E, Casado JA, Tischkowitz MD, Bueren JA, Creus A, Marcos R, et al. A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood 2005; 105(5):1946–9.
6. Kutler D, Auerbach A. Fanconi anemia in Ashkenazi Jews. Fam Cancer 2004; 3(3–4):241–8.
7. Morgan N V., Essop F, Demuth I, De Ravel T, Jansen S, Tischkowitz M, et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood 2005; 105(9):3542–4.
8. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009; 668(1–2):4–10.
9. Mikat B, Roll C, Schindler D, Gembruch U, Klempert I, Buiting K, et al. X-linked recessive VACTERL-H due to a mutation in FANCB in a preterm boy. Clin Dysmorphol 2016; 25(2):73–6.
10. Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: A report from the International Fanconi Anemia Registry. Pediatrics. 1993; 91(6):1116–20.
11. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol 2017; 177(4):526–42.
12. Alter BP, Rosenberg PS. VACTERL-H association and fanconi anemia. Mol Syndromol 2013; 4(1–2):87–93.
13. Clauson C, Schärer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA inter strand cross-link repair. Cold Spring Harb Perspect Biol 2013; 3(10):a012732.
14. Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol 2015; 37:49–60.
15. García-De-teresa B, Rodríguez A, Frias S. Chromosome instability in fanconi anemia: From breaks to phenotypic consequences. Genes (Basel) 2020; 11(12):1–35.
16. Bogliolo M, Surrallés J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33:32–40.
17. Che R, Zhang J, Nepal M, Han B, Fei P. Multifaceted Fanconi Anemia Signaling. Trends Genet 2018; 34(3):171–83.
18. Datta A, Brosh RM. Holding all the cards—how fanconi anemia proteins deal with replication stress and preserve genomic stability. Genes (Basel) 2019; 10(2):170.
19. Moreno OM, Paredes AC, Suarez-Obando F, Rojas A. An update on Fanconi anemia: Clinical, cytogenetic and molecular approaches (review). Biomed Reports 2021; 15(3):1–10.
20. Nepal M, Che R, Zhang J, Ma C, Fei P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017; 3(12):840-856.
21. Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet 2004; 36(11):1219–24.
22. Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: New players and new functions. Nat Rev Mol Cell Biol 2016; 17(6):337–49.
23. Peake JD, Noguchi E. Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum Genet 2022 May 21. doi: 10.1007/s00439-022-02462-9. Epub ahead of print.
24. Bogliolo M, Pujol R, Aza-Carmona M, Muñoz-Subirana N, Rodriguez-Santiago B, Casado JA, et al. Optimised molecular genetic diagnostics of Fanconi anaemia by whole exome sequencing and functional studies. J Med Genet 2020; 57(4):258- 268.
25. Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi Anemia Registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood 1989; 73(2):391–6.
26. Krausz C, Riera-Escamilla A, Chianese C, Moreno-Mendoza D, Ars E, Rajmil O, et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med 2019; 21(1):189–94.
27. Castella M, Pujol R, Callén E, Ramírez MJ, Casado JA, Talavera M, et al. Chromosome fragility in patients with Fanconi anaemia: Diagnostic implications and clinical impact. J Med Genet 2011; 48(4):242–50.
28. Moreno OM, Sánchez AI, Herreño A, Giraldo G, Suárez F, Prieto JC, et al. Phenotypic Characteristics and Copy Number Variants in a Cohort of Colombian Patients with VACTERL Association. Mol Syndromol 2020; 11:271–83.
29. Auerbach AD. Diagnosis of Fanconi Anemia by diepoxybutane analysis. Curr Protoc Hum Genet 2015; 2015:8.7.1-8.7.17.
30. Swift MR and Hirschhron K. FA’s Inherited Susceptibility to chromosome breakage. Ann Intern Med 1966; 65(3):496-503.
31. Auerbach AD, Wolman SR. Susceptibility of Fanconi’s anaemia fibroblasts to chromosome damage by carcinogens. Nature 1976; 261(5560):494–6.
32. Aslan D. Failure or delay in diagnosing Fanconi anemia - A welldefined genetic disorder. Turk J Pediatr 2013; 55(4):462–4.
33. Oostra AB, Nieuwint AWM, Joenje H, De Winter JP. Diagnosis of fanconi anemia: Chromosomal breakage analysis. Anemia 2012; 2012:238731.
34. Esmer C, Sánchez S, Ramos S, Molina B, Frias S, Carnevale A. DEB Test for Fanconi Anemia Detection in Patients with Atypical Phenotypes. Am J Med Genet 2004; 124A(1):35–9.
35. Reutter H, Ludwig M. VATER/VACTERL association: Evidence for the role of genetic factors. Mol Syndromol 2013; 4(1–2):16–9.
36. Trujillo JP, Surralles J. Savior siblings and Fanconi anemia: analysis of success rates from the family’s perspective. Genet Med 2015; 17:935–8.
37. Korgaonkar S, Ghosh K, Jijina F, Vundinti BR. Chromosomal breakage study in children suspected with fanconi anemia in the indian population. J Pediatr Hematol Oncol 2010; 32(8):606– 10.
38. Cirkovic S, Guc-Scekic M, Vujic D, Ilic N, Micic D, Skoric D, et al. Diagnosis of Fanconi’s anemia by diepoxybutane analysis in children from Serbia. Balk J Med Genet 2011; 14(2):65–70.
39. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010; 24(3):101–22.
40. Zierhut HA, Tryon R, Sanborn EM. Genetic Counseling for Fanconi Anemia: Crosslinking Disciplines. J Genet Couns 2014; 23(6):910–21.
41. Auerbach AD. A test for Fanconi’s anemia. Blood 1988; 72(1):366– 9.
42. Fargo JH, Rochowski A, Giri N, Savage SA, Olson SB, Alter BP. Comparison of chromosome breakage in non-mosaic and mosaic patients with Fanconi anemia, relatives, and patients with other inherited bone marrow failure syndromes. Cytogenet Genome Res 2014; 144(1):15–27.
43. Nicoletti E, Rao G, Bueren JA, Río P, Navarro S, Surrallés J, et al. Mosaicism in Fanconi anemia: concise review and evaluation of published cases with focus on clinical course of blood count normalization. Ann Hematol 2020; 99:913–24.
44. Revy P, Kannengiesser C, Fischer A. Somatic genetic rescue in Mendelian haematopoietic diseases. Nat Rev Genet 2019; 20(10):582–98.
45. Waisfisz Q, Morgan N V., Savino M, De Winter JP, Van Berkel CGM, Hoatlin ME, et al. Spontaneous functional correction of homozygous Fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet 1999; 22(4):379–83.
46. Soulier J, Soulier J, Leblanc T, Larghero J, Dastot H, Shimamura A, Guardiola P, et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/ BRCA pathway. Blood 2005 ; 105(3):1329–36.
47. Mankad A, Taniguchi T, Cox B, Akkari Y, Rathbun RK, Lucas L, et al. Natural gene therapy in monozygotic twins with Fanconi anemia. Blood 2006; 107(8):3084–90.
48. Ramírez MJ, Pujol R, Trujillo-Quintero JP, Minguillon J, Bogliolo M, Rio P, et al. Natural gene therapy by reverse mosaicism leads to improved hematology in Fanconi anemia patients. Am J Hematol 2021; 96:989–999.
49. Hyde RK, Liu PP. GATA2 mutations lead to MDS and AML. Nat Genet 2011; 43(10):926–7.
50. Sarkar K, Han SS, Wen KK, Ochs HD, Dupré L, Seidman MM, et al. R-loops cause genomic instability in T helper lymphocytes from patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2018; 142(1):219–34.
51. Mehta PA, Harris RE, Davies SM, Kim MO, Mueller R, Lampkin B, et al. Numerical chromosomal changes and risk of development of myelodysplastic syndrome-acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet Cytogenet 2010; 203(2):180–6.
52. Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anemia: An International Fanconi Anemia Registry study. Blood 1994; 84(5):1650–5.
53. Cioc Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: Morphologic and cytogenetic characteristics. Am J Clin Pathol 2010; 133(1):92–100.
54. Rochowski A, Olson SB, Alonzo TA, Gerbing RB, Lange BJ, Alter BP. Patients with Fanconi anemia and AML have different cytogenetic clones than de novo cases of AML. Pediatr Blood Cancer 2012; 59(5):922–4.
55. Taylor AMR, Rothblum-Oviatt C, Ellis NA, Hickson ID, Meyer S, Crawford TO, et al. Chromosome instability syndromes. Nat Rev Dis Primers 2019; 19;5(1):64.
56. Vásquez Palacio G, Ramírez Castro JL, Posada Díaz Á, Sierra M, Botero OL, Durango NE, et al. Leucemia linfoide aguda: Estudio citogenético en niños atendidos en el Hospital Universitario San Vicente de Paúl de Medellín en el período 1998-2001. Iatreia 2002; 15(4):217–25.
57. Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003; 101(4):1249–56.
58. Fiesco-roa MO, Giri N, Mcreynolds LJ, Best AF, Alter BP. Blood Reviews Genotype-phenotype associations in Fanconi anemia : A literature review. Blood Rev 2019; 37:100589.
59. Smith AR, Wagner JE. Current clinical management of Fanconi anemia. Expert Rev Hematol. 2012;5(5):513–22.
60. Giri N, Batista DL, Batista DL, Alter BP, Stratakis CA. Endocrine Abnormalities in Patients with Fanconi Anemia. J Clin Endocrinol Metab 2007; 92(7):2624-31.
61. Huck Hanenberg H, Gudowius S, Fenk R, Kalb R, Neveling K, et al. Delayed diagnosis and complications of Fanconi anaemia at advanced age - A paradigm. Br J Haematol 2006; 133(2):188–97.