Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v55i1.264
Revisión
Características infrecuentes del síndrome de Miller-Fisher. Revisión de la literatura a propósito de un caso.
Uncommon Features of Miller-Fisher Syndrome. Case Report and review of the literature
Ruth Camila Púa Torrejóna, Amanda Bermejo Gómeza, Ana Gómez-Carpintero Garcíaa, Rebeca Villares Alonsoa.
a. Hospital Universitario de Móstoles. Madrid, España.
Recibido 17 de enero de 2021 Aceptado 20 de mayo de 2022
Como Citar: Púa Torrejón RC, Bermejo Gómez A, Gómez-Carpintero García A, Villares Alonso R. Características infrecuentes del síndrome de Miller-Fisher. Revisión de la literatura a propósito de un caso. Pediatr. 2022;55(1:)30-35.
Autor para correspondencia: Ruth Camila Púa Torrejón.
Correo electrónico: ruth_kapry@hotmail.com
Editor adjunto: Alvaro León Jácome Orozco
Resumen
Antecedentes: el síndrome de Miller Fisher es una polineuropatía autoinmune aguda, caracterizada por la triada clínica de arreflexia, ataxia y oftalmoplejía. Es una patología infrecuente en la edad pediátrica que se asocia, en la mayoría de los casos, a un antecedente de proceso infeccioso. En el análisis del líquido cefalorraquídeo destaca la disociación albúmino citológica y la presencia de anticuerpos IgG antigangliósido contra GQ1b en más del 80% de los casos. Reporte de caso: niña de 5 años con la triada clínica características, de curso agudo y antecedente de infección gastrointestinal (sin agente infeccioso conocido). En el líquido cefalorraquídeo se describen hallazgos poco frecuentes en este síndrome como son leucocitosis y positividad de anticuerpos IgG antigangliósido contra GT1a. Los demás anticuerpos antigangliósido fueron negativos. Las pruebas complementarias no son útiles para confirmar o descartar el diagnóstico, ya que este es principalmente clínico, sin embargo, sirven para descartar otras patologías incluidas en el diagnostico diferencial. Conclusión: es necesario un diagnóstico precoz para establecer las medidas de soporte adecuadas. A pesar de que las inmunoglobulinas y la plasmaféresis han sido comúnmente utilizadas para su tratamiento, son necesarios experimentos clínicos aleatorizados que demuestren su eficacia.
Palabras clave: Anticuerpos antigangliósido, arreflexia, ataxia, oftalmoplejía, Síndrome de Miller Fisher.
Abstract
Background: Miller Fisher syndrome is an acute autoimmune polyneuropathy characterized by the clinical triad of areflexia, ataxia, and ophthalmoplegia. It is a rare pathology in the pediatric age that is associated, in most cases, with a history of an infectious process. In the cerebrospinal fluid analysis, cytological albumin dissociation and IgG antiganglioside antibodies against GQ1b stand out in more than 80% of cases. Case report: 5-year-old girl with the characteristic clinical triad, acute course, and history of gastrointestinal infection (no known infectious agent). Uncommon findings in the cerebrospinal fluid in this syndrome are described, such as leukocytosis and positivity for IgG antiganglioside antibodies against GT1a. The other antiganglioside antibodies were negative. The complementary tests are not helpful in confirming or ruling out the diagnosis since this is mainly clinical; however, they serve to rule out other pathologies included in the differential diagnosis. an early diagnosis is necessary to establish adequate support measures. Although immunoglobulins and plasmapheresis have been commonly used for their treatment, randomized clinical trials are needed to demonstrate their efficacy.
Key words: Antiganglioside antibodies, arreflexia, ataxia, Miller Fisher Syndrome, ophthalmoplegia.
Introducción
El síndrome de Miller Fisher (SMF) es una polineuropatía desmielinizante, autoinmune y aguda (1). Se estima una incidencia aproximada de 1 a 2 por cada 1 000 000 habitantes por año y presenta un pico de incidencia en primavera, con predominio en varones, con una relación hombre mujer de aproximadamente 2:1 (2- 4).
A pesar de ser una enfermedad rara, es considerada la variante de presentación más frecuente del síndrome de Guillain-Barré (SGB) (1-3). Algunos estudios sugieren que forma parte de un espectro continuo de trastornos autoinmunes que pueden afectar al sistema nervioso periférico o central, describiendo casos de SMF que evolucionan a SGB (1-3).
A su vez, existen diferentes subtipos dentro del SMF: clásico (variante más frecuente), neuropatía ataxia aguda, encefalitis de Bickerstaf e hipersomnolencia atáxica aguda entre otras, pudiendo existir superposición entre ellas (2,4). Estas entidades comparten una misma etiología y mecanismo fisiopatológico con diferentes manifestaciones clínicas.
Si bien, muchos autores consideran la encefalitis de Bickerstaf como un posible continuum del Síndrome de Miller Fisher, nosológicamente son dos entidades diferentes. El síndrome de Miller Fisher afecta al sistema nervioso periférico y la encefalitis es del sistema nervioso central, clínicamente la encefalitis de Bickerstaf cursa con alteración de conciencia, mientras que el síndrome de Miller Fisher no. Además, las neuroimágenes son diferentes. Por todo lo anterior, si bien son cuadros similares, se trata de dos entidades clínicamente diferentes, pero con una base inmunológica común (1,3,5).
El SMF se asocia, en la mayoría de los casos, a un antecedente de proceso infeccioso sobre todo del tracto respiratorio superior y en menor porcentaje a un proceso infeccioso de origen digestivo, con una duración media de siete días (intervalo 1 a 26 días) y una media entre la infección y el inicio de la sintomatología neurológica de ocho días (intervalo 1 a 42 días) (1-3,6).
Se estima que, en un 18 % de los casos de SMF, este se presenta sin síntomas infecciosos previos al inicio de la clínica neurológica (3). Los agentes infecciosos más frecuentemente relacionados son Campylobacter jejuni y Haemophilus influenzae, seguido en orden de frecuencia por Citomegalovirus, Mycoplasma pneumoniae y virus Epstein-Barr, entre otros (2,3,6). En el 56 al 67 % de los casos no se ha identificado el agente infeccioso implicado a pesar de que en un 82 al 94 %, los pacientes tenían algún síntoma de infección, lo que apoyaría la necesidad de realizar más investigaciones en este ámbito (2,6,7).
El diagnóstico se basa principalmente en la sospecha clínica, caracterizada por la triada clínica de oftalmoplejía, ataxia y arreflexia con un curso monofásico (1-4,7). El síntoma de inicio del SMF más frecuente es la diplopía seguido de la ataxia. Otros síntomas de presentación descritos son las disestesias en extremidades inferiores, disfagia, blefaroptosis o fotofobia (1-4,6). Otros síntomas frecuentes que se pueden encontrar durante el curso de la enfermedad son la midriasis y la parálisis facial periférica; mientras que la perdida de sensibilidad, alteraciones miccionales, somnolencia, disgeusia y la debilidad son infrecuentes (3,6).
Aunque el diagnostico se basa en la clínica, el análisis del líquido cefalorraquídeo (LCR), los estudios de conducción nerviosa, las pruebas de imagen y las pruebas serológicas pueden ayudar a confirmar el diagnóstico [4]. La característica principal del LCR es la disociación albumino citológica que puede estar ausente los primeros días del cuadro y aparecer posteriormente. En una serie de 375 pacientes se observó́ disociación albumino citológica en el 37 % de los casos en la primera semana de evolución, ascendiendo hasta el 76 % en la segunda semana y siendo máxima a las 4 a 6 semanas.
La pleocitosis se objetivó en un 4 % de los casos en la primera semana y en un 5 % en la segunda semana, con una mediana de leucocitos de 1 célula/μl (intervalo 1 - 105) y 2 célula/μl (intervalo 0 - 42) respectivamente (siendo encontrado más frecuentemente en la encefalitis de Bickerstaff) (2).
Los estudios de conducción nerviosa demuestran la afectación desmielinizante y en el estudio electromiográfico (EMG), se suele observar una disminución de la amplitud de los potenciales de acción musculares, enlentecimiento de la velocidad de conducción y ausencia de reflejos H del sóleo (2).
En algunos estudios se ha objetivado un 1 % de resultados anormales en forma de aumento de la intensidad de señal en la resonancia nuclear magnética (RNM) de estos pacientes, siendo las localizaciones afectadas el mesencéfalo, cerebelo y el pedúnculo cerebral medio (2).
Muchos pacientes presentan elevación de anticuerpos IgG antigangliósidos contra GQ1b en la fase aguda de la enfermedad, llegando en algunas series hasta el 83 al 89 % de los casos (1-4,7). Estos anticuerpos no están presentes en personas sanas (2). Estos anticuerpos antigangliósidos no son exclusivos del SMF, pudiendo estar presentes en otras entidades con la encefalitis de Bickerstaf (8). Las asociaciones clínico-serológicas se describen en la tabla 1.
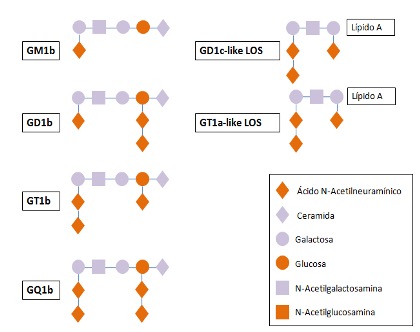
Los gangliósidos, componentes importantes de los nervios periféricos, están compuestos por una ceramida (N-aciladaesfingosina) unida a uno o más azucares (hexosas) y contienen un núcleo de oligosacárido enlazado al ácido siálico. Los principales gangliósidos descritos son: GQ1b, GT1a, GD1a y GM1, que difieren en el número (Q: cuatro, T: tres, D: dos, M: uno) y posición del ácido siálico (8). La localización de estos gangliósidos se asocia a las manifestaciones clínicas; así́, por ejemplo, el gangliósido GQ1b es fuertemente expresado en nervios oculomotor, troclear y abducens y en los husos musculares, dando lugar a la oftalmoplejía y arreflexia; los nervios glosofaríngeo y vago expresan, principalmente, GT1a y GQ1b (1). Los anticuerpos GQ1b tienen reactividad cruzada con los anticuerpos GT1a (3).
Los lipooligosacáridos (LOS) son la estructura principal de la superficie celular de algunos patógenos implicados en el SMF como el Campylobacter jejuni o el Haemophilus influenzae (2). La sialiltransferasa bacteriana Cst-II actúa en la biosíntesis de estos LOS de tipo gangliósido, determinando su actividad enzimática en el aminoácido 51. La sialiltransferasaCst-II (Asn 51) produce GT1a-like y GD1c-like lipooligosacáridos en el que cada trisacárido terminal es idéntico al GQ1b (2). La estructura química de los oligosacáridos expresados en la membrana de los microorganismos es muy similar a la de los gangliósidos humanos (Figura 1), y, por «mimetismo molecular», estimula a células B a producir anticuerpos que se fijan sobre estos gangliósidos que se encuentran ubicados en la periferia de los nodos de Ranvier (2,8). Esto provoca activación de complemento, formación del complejo de ataque de membrana y disrupción de los canales de sodio dependientes de voltaje, haciendo que la conducción nerviosa se vea afectada con la consiguiente aparición de bloqueos de conducción y entrada de macrófagos al espacio periaxonal, dañando los axones (8).
Son muchas las evidencias científicas que apoyan el mimetismo molecular existente ente los gangliósidos GQ1b, GT1a, GM1b, GD1a y los epítopos del lipopoligosacárido del C. jejuni y H. influenzae (2,3,7). Estos epítopos no se han detectado en algunas cepas de C. jejuni presentes en algún caso de SMF, lo que indicaría que otros mecanismos fisiopatológicos estarían implicados (7).
Inicialmente se produce la infección por un microorganismo que contiene el epítopo GQ1b en el lipooligosacárido de su superficie, induciendo la producción de anticuerpos antiGQ1b en determinados pacientes. Posteriormente estos anticuerpos se unen a los gangliósidos GQ1b expresados en los nervios oculomotores y las neuronas sensoriales primarias induciendo el SMF (2). Si estos anticuerpos antiGQ1b atraviesan la barrera hematoencefálica se produciría la encefalitis de Bickerstaff al unirse a los GQ1b de la formación reticular del tronco cerebral (1,2).
La presencia de estos anticuerpos en cuadros clínicos dudosos podría ayudar a confirmar el diagnóstico, pero su negatividad no lo descarta y no se debería posponer el inicio del tratamiento en los casos indicados a la espera de los resultados. Títulos elevados y persistentes podrían estar asociados a un peor pronóstico de la enfermedad (8).
En general, se trata de una enfermedad con buen pronóstico, siendo raras las recurrencias (5 al 10 %) (3,4). La tasa de mortalidad es inferior al 5 % (4). La arreflexia es el síntoma más tardío en resolverse. Desde la aparición hasta el comienzo de la recuperación de la ataxia y la oftalmoplejía transcurren una media de 12 a 15 días respectivamente y de 32 a 88 días respectivamente hasta la recuperación completa (3,4).
La mayoría no tienen grandes discapacidades y vuelven a realizar sus actividades habituales a los 6 meses del comienzo. Raramente se complica con insuficiencia respiratoria con necesidad de soporte ventilatorio (3,5).
Los factores asociados a un peor pronóstico serían la progresión rápida de la enfermedad, la necesidad de soporte respiratorio y la evidencia electrofisiológica de degeneración axonal; no interfiriendo en el pronóstico el sexo, la evidencia de infección previa, la discapacidad en el pico de la enfermedad ni la latencia hasta el pico de la enfermedad (3). Numerosos trabajos demostraron la vinculación entre una recuperación clínica incompleta y la presencia de anticuerpos antigangliósidos circulantes (8).
El manejo del SMF requiere hospitalización para vigilancia de las posibles, aunque raras, complicaciones respiratorias y digestivas. Debido al curso favorable de la enfermedad ha sido discutido en numerosas ocasiones la necesidad de asociar un tratamiento específico al tratamiento de soporte (3,9).
Ningún experimento clínico aleatorizado ha demostrado la eficacia de la plasmaféresis o las inmunoglobulinas intravenosas en pacientes con SMF, aunque como variante de SGB han sido aplicados estos tratamientos (4,5,9). En estudios observacionales se ha objetivado leve aceleración en la resolución de la oftalmoplejía y la ataxia, pero el tiempo hasta la recuperación completa y la mortalidad fue similar entre los grupos tratados con inmunoglobulinas o plasmaféresis y el grupo control (4,5,9).
Algunos estudios indican que el tratamiento con inmunoglobulinas o plasmaféresis podría estar justificado en aquellos pacientes con disfagia o dificultad respiratoria a pesar de no estar demostrada su eficacia (4,5,9). La elección entre uno u otro tratamiento vendría establecida por el hecho de que las inmunoglobulinas están más disponibles y tienen menos efectos adversos (10).
Las inmunoglobulinas tienen efectos inmunomoduladores pleiotrópicos, incluidos la neutralización de anticuerpos y citoquinas, y la inhibición de la activación del complemento. Su farmacocinética ha sido evaluada en controles sanos y pacientes, mostrando una considerable variabilidad. La dosis óptima de inmunoglobulinas es desconocida. La dosis estándar actual de 2 g/Kg se ha establecido de manera arbitraria y no ha sido definida en estudios de búsqueda de dosis por subgrupos, por lo que, si la inmunomodulación dependiese de la dosis, una baja concentración de IgG supondría una supresión inmune subóptima, con la consecuente prolongación y extensión del daño y unos peores resultados; por lo tanto, son necesarios más estudios en este ámbito (10).
La terapia física y la rehabilitación es importante para prevenir contracturas y mantener la función del aparato locomotor. Se debe establecer un plan individual con el objetivo de aumentar la fuerza muscular, reducir el dolor y evitar complicaciones (4).
Tabla 1. Entidades clínicas del SGB y anticuerpos más frecuentemente relacionados [8].
|
Entidades clínicas |
Anticuerpos frecuentemente relacionados |
|
Neuropatía axonal motora y sensitiva |
GM1, GM1b, GD1a |
|
Neuropatía axonal motora aguda |
GM1, GM1b, GD1A, GalNac-GD1a |
|
Neuropatía sensitiva aguda |
GD1b |
|
Síndrome de Miller-Fisher |
GQ1b, GT1a |
|
Variante faringobraquial |
GT1a |
|
Síndrome de superposición Miller-Fisher/Guillain-Barré |
GQ1b, GM1, GM1b, GD1a, GalNac-GD1a |
Figura 1. Estructura de los gangliósidos GM1b, GD1b, GT1a, GQ1b y lipooligosacáridos GT1a-like LOS y GD1c-like LOS [2,8]
Reporte de caso
Niña de cinco años que acude al servicio de urgencias de un hospital de segundo nivel por cuadro de diplopía binocular en visión lejana de ocho horas de evolución, seguido de inestabilidad de la marcha de inicio brusco.
Aportó una determinación de tóxicos en orina negativa y tomografía axial computarizada (TAC) craneal normal, realizada en otro hospital de tercer nivel cuatro horas antes de su llegada al centro.
Se describen episodios de deposiciones de consistencia disminuida con mucosidad, sin otros productos patológicos, desde diez días antes de su consulta a urgencias y resuelta alrededor de 12 horas previas al ingreso. Perdida de 3 Kg de peso. Asociaba dolor en extremidades inferiores de 48 horas de evolución sin antecedente de traumatismo ni esfuerzo físico.
Como antecedentes, la paciente fue producto de embarazo normal. Parto eutócico a término con peso adecuado sin incidencias en periodo neonatal y cribado de enfermedades endocrino- metabólicas negativo. Desarrollo psicomotor normal sin enfermedades previas de interés. Vacunada según calendario de la Comunidad de Madrid (España). No realizó viajes recientes. Mantiene dos jerbos en domicilio desde 5 días previos al inicio de la sintomatología. No antecedentes familiares de interés.
A la exploración física a su llegada, con constantes normales, diplopía binocular en todas las posiciones de la mirada que desaparece a la visión monocular, ptosis bilateral leve y limitación de la abducción de ambos ojos de predominio derecho. Pupilas isocóricas y normo reactivas sin nistagmo. Fondo de ojo bilateral normal. Fuerza conservada. Arreflexia en extremidades inferiores a su llegada, que asciende progresivamente con arreflexia universal a las 18 horas de su arribo. Dismetría y tándem inestable con aumento de la base de sustentación. Resto de exploración física general y neurológica normal.
Se realiza analítica con hemograma y bioquímica normales con PCR 0.4 mg/L y aumento a las 48 horas hasta 12.2 mg/L y VSG 50 mm/h. Ante sospecha de SMF (oftalmoplejía, ataxia y arreflexia) se administra dosis de inmunoglobulinas intravenosas a dosis estándar de 2g/Kg a las 19 horas de su llegada a urgencias. Los valores de las inmunoglobulinas se analizaron al tercer día de ingreso, tras administración de inmunoglobulinas, estando elevada la IgA, IgM y la IgG (esta última más de dos veces el valor normal).
Al segundo día de ingreso se realizó una punción lumbar que muestra una presión normal del líquido cefalorraquídeo (LCR), pleocitosis (94/uL) con predominio de polimorfonucleares (63 %) y resto de parámetros normales. PCR para virus y cultivo de bacterias y hongos resultan posteriormente negativos. Se envía muestra para estudio de anticuerpos antigangliósidos resultando los anticuerpos GT1a tipo IgG positivos, con resto de anticuerpos negativos, lo que apoyó el diagnóstico de sospecha. Se repite punción lumbar a la semana de ingreso objetivándose disociación albumino-citológica (215 mg/ dl de proteínas y 6/uL leucocitos) con PCR de virus y cultivos de negativos.
Para completar estudio, se realizan sistemático de orina sin datos de infección y cultivo de heces que resulta negativo. El electromiograma y la neurocoducción (NC), a los 4 días de ingreso, mostró datos sugerentes de desmielinización aguda con afectación parcial de extremidades inferiores y electroencefalograma con alteraciones inespecíficas durante el sueño. La RMN cerebral no reveló alteraciones significativas.
Permaneció ingresada en planta de hospitalización bajo vigilancia estrecha durante 15 días, presentando decaimiento, somnolencia y episodios de cefaleas ocasionales los primeros días de ingreso, sin disnea ni disfagia. Tras administración de la dosis de inmunoglobulina intravenosa presenta mejoría progresiva sin resolución completa de la ataxia y la oftalmoplejía, persistiendo arreflexia universal al alta.
Se realizaron revisiones en consultas de Neuropediatría, siendo la marcha normal sin dismetría ni disdiadococinesia al mes, pudiendo realizar actividades deportivas. Presenta, además, desde el ingreso, dolor en extremidades inferiores que precisa analgesia pautada con antiinflamatorios no esteroideos y gabapentina, que es posible retirar completamente a los 2 meses del alta. A los cinco meses la diplopía ya era intermitente, persistiendo la arreflexia universal que se mantiene en revisión al año; así como, episodios de caídas frecuentes.
Discusión
El SMF es una enfermedad poco frecuente, considerada incluso enfermedad rara, no habiéndose notificado casos en menores en algunos países. Se estima una incidencia de 1 a 2 por cada 1 000 000 de habitantes año en adultos, siendo muy limitados los datos epidemiológicos en la edad pediátrica.
En el caso presentado existía un antecedente infeccioso con clínica gastrointestinal iniciado en los diez días previos a la clínica neurológica, no pudiéndose hallar el agente infeccioso que, por la clínica descrita, podría haberse tratado de una gastroenteritis por Campylobacter jejuni, agente infeccioso más comúnmente implicado en la literatura en los SMF con antecedente de gastroenteritis infecciosa.
El tiempo entre la infección y el inicio de la clínica neurológica y la duración de esta infección fue el habitualmente descrito en la literatura. En cuanto a la clínica, la paciente manifestó la clínica típica de manera progresiva, siendo la oftalmoplejía el síntoma de inicio con limitaciones del VI par bilateral y ptosis bilateral (III par) sin limitaciones en el resto de los movimientos oculomotores (IV par indemne). El síntoma de inicio más frecuente, en la mayoría de las series publicadas, es la oftalmoplejía bilateral y con afectación de los tres nervios oculomotores.
A pesar de que el diagnóstico es clínico, se decidió́ realizar pruebas complementarias para apoyar el diagnóstico. El análisis del LCR reveló leucocitosis, hallazgo ausente en el 96 % de los pacientes con SMF y frecuente en la encefalitis de Bickerstaf; si bien, hay que tener en cuenta que dicho análisis de LCR se realizó posteriormente a la administración de inmunoglobulinas intravenosas no pudiéndose demostrar si la leucocitosis es secundaria a las mismas o al SMF en sí. La disociación albúmino citológica no apareció hasta la segunda punción lumbar realizada al 7º día de ingreso. Se obtuvo positividad de los anticuerpos antigangliósidos GT1a en LCR. Sin embargo, los anticuerpos antiGQ1b, que son positivos en el 83 al 89 % de los casos de SMF, resultaron negativos. Se sabe de la existencia de reactividad cruzada entre los anticuerpos antiGQ1b y antiGT1a. La negatividad de dichos anticuerpos no excluye el diagnóstico.
Se ha propuesto que la sintomatología neurológica depende de la localización de estos gangliósidos, lo que podría utilizarse como factor pronostico. Estos anticuerpos antiGT1a se expresan predominantemente en los nervios glosofaríngeo y vago, sin embargo, nuestra paciente no presentó disfagia ni clínica respiratoria.
La positividad de estos anticuerpos respaldaría la presencia de un agente infeccioso con el epítopo GT1a en el lipooligosacárido de su superficie, entre ellos Campylobacter jejuni, que indujese la producción de anticuerpos antiGT1a y por mimetismo molecular se uniese a los gangliósidos expresados en los nervios, husos musculares, tracto espinocerebeloso y sistema propioceptivo produciendo la triada característica del SMF.
El resto de las pruebas complementarias fueron compatibles con los hallazgos típicos del SMF: patrón desmielinizante en EMG y NC y normalidad de EEG y RNM, permitiendo descartar diagnósticos alternativos.
Teniendo en cuenta lo anterior el presente caso tendría componente del SMF y de la encefalitis de Bickerstaf, esta última por la presencia de leucocitosis en el LCR y la alteración del nivel de conciencia.
La paciente recibió medidas de soporte y tratamiento sintomático. Ante la sospecha, se inició tratamiento con inmunoglobulinas a dosis estándar. Se prefirió frente a la plasmaféresis por su mayor disponibilidad, menores efectos adversos y mayor facilidad de administración. Sin embargo, los datos relativos a la eficacia del tratamiento del SMF obtenidos a través de ensayos clínicos son escasos y los obtenidos a través de estudios observacionales demuestran nulo o escaso beneficio, acelerando la mejoría de la oftalmoplejía y la ataxia, siendo necesarios más estudios en este ámbito.
La evolución de la paciente siguió un curso benigno típico, pudiendo ser dada de alta a los quince días de ingreso. La ataxia y la oftalmoplejía mejoraron a los dos días tras la administración de las inmunoglobulinas. Durante su ingreso, presentó otros síntomas descritos en otros pacientes con SMF como disestesias, decaimiento, somnolencia y cefalea, sin presentar disfagia ni dificultad respiratoria que precisase soporte respiratorio. Lo que podría considerarse atípico es la persistencia de la arreflexia y las caídas frecuentes un año tras el debut del síndrome.
Ante un paciente que presente la tríada típica del síndrome de Miller-Fisher pero que durante su evolución presenta alteración del nivel de conciencia hay que pensar en una encefalitis de Bickerstaf sin olvidar la posible superposición entre ambas entidades.
Bibliografía
1. Wakerley B, Uncini A, Yuki N. Guillain-Barré and Miller Fisher syndromes-new diagnostic classification. Nat. Rev.Neurol 2014; 10(9):537-44.
2. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K, et al. Bickerstaff’s brainstem encephalitis and Fisher sindrome form a continuous spectrum. Clinical analysis of 581 cases. J Neurol 2008; 255:674-682.
3. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher sindrome. Neurology 2001; 56:1104-6.
4. Al Othman B, Raabe J, Kini A, Lee, A. Up to date: the Miller Fisher variants of Guillain-Barré syndrome. Curr Opin Ophthalmol 2019; 30(6):462-6.
5. Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher syndrome. Neurology2007; 68:1144–1146.
6. Koga M, Kishi M, Fukusako T, Ikuta N, Kato M, Kanda T. Antecedent infection in Fisher syndrome: sources of variation in clinical characteristics. Journal of Neurology 2019; 266:1655- 1662.
7. Koga M, Gilbert M, Li J. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology 2005; 64:1605-11.
8. Reisin R, Salutto V, Aguirre F, Álvarez V, Barroso F, Bendersky M, et al. Utilidad de la identificación de anticuerpos en neuropatías periféricas, neuropatías y gangliopatías: revisión. NeurolArg 2020; 12(2): 98-112.
9. A van Doorn P, Ruts L, Jacobs B. Clinical features, pathogenesis, and treatment of Guillain- Barré syndrome. Lancet Neurology 2008; 7(10):939-50.
10. Kuitwaard K, de Gelder J, Tio‐Gillen A, Hop W, van Gelder T, van Toorenenbergen A, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann. Neurol 2009; 66(5):597-603.
