Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v55i4.187
Reporte de caso
Mucopolisacaridosis tipo I, variante síndrome de Hurler: Abordaje inicial y relación con la literatura
Mucopolysaccharidosis type I, Hurler syndrome variant : Initial approach and relationship to literature
Sergio Vergaraa, María Paula Prietoa, Oriana Lujána, Lorena Rinconesa, Maria Ximena Arteagaa, Natalia Gómeza, Andreina Zannina, Amenaida Ferrera, Isabel Fernándezb, Luis Gustavo Celisa.
a. Facultad de Medicina – Universidad de La Sabana, Bogotá, Colombia.
b. Unidad de Genética Médica. Policlínica Metropolitana, Caracas, Venezuela
Recibido 01 de abril de 2020 Aceptado 08 de noviembre de 2022
Como Citar: Vergara S, Prieto MP, Luján O, Rincones L, Arteaga MX, Gómez N, Zannin A, Ferrer A, Fernández I, Celis LG. Mucopolisacaridosis tipo
I, variante síndrome de Hurler: Abordaje inicial y relación con la literatura. Pediatr. 2022;55(3): 209-214.
Autor para correspondencia: Sergio Vergara
Correo electrónico: sergiovercar@unisabana.edu.co
Editor jefe: Fernando Suárez-Obando
Resumen
Antecedentes: las enfermedades de depósito tipo mucopolisacaridosis son un grupo de enfermedades genéticas poco frecuentes con patrón de herencia tipo autosómico recesivo. La mucopolisacaridosis (MPS) es considerada específicamente una condición de sobrecarga lisosomal causada por deficiencias de enzimas encargadas de la degradación de glicosaminoglicanos (GAG), también llamados mucopolisacáridos; este déficit enzimático se genera de la acumulación progresiva de compuestos en diferentes tejidos que conlleva a daño tisular generalizado y que tiende a progresar a falla multiorgánica (1–5). Reporte de caso: paciente femenina lactante mayor con retraso en el neurodesarrollo y alteraciones fenotípicas notorias, lo cual se relaciona con hallazgos descritos en la literatura. Conclusiones: se identificó déficit de la enzima alfa–L-iduronidasa, en contexto de un cuadro clínico con manifestaciones severas y la edad tan temprana de inicio de la patología, se cataloga dentro de la MPS I, Síndrome de Hurler. El avance en el abordaje temprano y conocimiento en la historia natural de las enfermedades de depósito permitirá generar un mejor abordaje diagnóstico y terapéutico, generando un mejor desenlace.
Palabras clave: Anomalía cromosómica, alteración en el neurodesarrollo, Mucopolisacaridosis tipo 1, enfermedad de depósito lisosomal, síndrome de Hurler.
Abstract
Background: Mucopolysaccharidoses-type storage diseases are a group of rare genetic diseases with an autosomal recessive inheritance pattern. Mucopolysaccharidosis (MPS) is a condition of lysosomal overload caused by deficiencies of enzymes responsible for the degradation of glycosaminoglycans (GAG), also called mucopolysaccharides. This enzyme deficiency is generated from the progressive accumulation of compounds in different tissues that leads to generalized tissue damage and tends to progress to multiorgan failure (1–5). Case report: Elderly lactating female patient with neurodevelopmental delay and notable phenotypic alterations, which is related to findings described in the literature. Conclusions: alpha-L-Iduronidase enzyme deficiency was identified in the context of a clinical picture with severe manifestations and such an early age of onset of the pathology, it is classified as MPS I, or Hurler Syndrome.Advances in the early approach and knowledge of the natural history of deposit diseases will make it possible to generate a better diagnostic and therapeutic approach, generating a better outcome.
Key words: Chromosomal Abnormality, Hurler Syndrome, l|ysosomal Storage Diseases, Mucopolysaccharidosis Type I, neurodevelopmental, disorders.
Introducción
La mucopolisacaridosis (MPS) es una enfermedad de depósito, específicamente de sobrecarga lisosomal causada por deficiencias de enzimas encargadas de la degradación de glicosaminoglicanos (GAG) o mucopolisacáridos, lo que ocasiona una acumulación progresiva de estos compuestos en diferentes órganos, lo que conlleva a un daño tisular generalizado y falla multiorgánica (2–5). Los GAG son hidratos de carbono complejos de cadena larga, compuestos por ácidos urónicos, amino azúcares neutros, dentro de los cuales se encuentran con mayor importancia los condroitín-4-sulfato, condroitín-6-sulfato, heparán sulfato, dermatán sulfato, queratán sulfato y hialuronano (6,7).
Esta enfermedad de depósito tiene diferentes presentaciones que se diferencian por el compromiso genético, la vía metabólica afectada y el sustrato acumulado (4). En la literatura se han descrito las siguientes variantes (3,5): 1) Síndrome de Hurler-Scheie o MPS I (OMIM 252800) la cual se divide a su vez en tres tipos, MPS IH (Hurler) siendo la forma más severa, MPS IHS (Hurler-Scheie) intermedia y MPS I S (Scheie) leve, esta última se conocía anteriormente como MPS V (6,8). 2) Síndrome de Hunter o MPS II. 3) Síndrome de San Filippo o MPS III (con los subtipos A, B, C y D). 4) Síndrome de Morquio o MPS IV (subtipos A y B). 5) Síndrome de Maroteaux-Lamy o MPS VI. 5) Síndrome de Sly o MPS VII y 6) Déficit de Hialuronidasa o y MPS IX La mayoría de las presentaciones de MPS presentan un patrón de herencia de tipo autosómico recesivo, sin embargo, MPS II es una enfermedad genética ligada al cromosoma X (5).
Se considera que las MPS son entidades raras, sin embargo, se ha registrado una incidencia general de todos los tipos de mucopolisacaridosis de aproximadamente 1 por cada 20.000 nacimientos y específicamente de MPS tipo I de 1 por cada 100 000 nacimientos. No obstante, la literatura documenta que existe actualmente un sub diagnóstico de esta patología debido a la falta de evidencia (4,7). Estudios indican que la mediana de supervivencia con dicha enfermedad es de 11.6 años (1).
El objetivo de este artículo es reportar un caso de una paciente pediátrica proveniente de Caracas, Venezuela con Mucopolisacaridosis tipo I, tipo síndrome de Hurler y hacer un análisis de la bibliografía reciente de la patología.
Reporte de caso
Se presenta una paciente femenina de 22 meses de edad, al momento del diagnóstico, proveniente de Caracas, Venezuela. Fruto de primera gestación de madre adolescente con antecedentes personales y familiares desconocidos, quien es llevada a consulta de genética en la Policlínica Metropolitana de Caracas, Venezuela.
Los padres refieren cuadro clínico que inicia a los seis meses de vida donde comienzan a notar desviación de la columna lumbar con posterior prominencia frontal y retraso en el desarrollo del lenguaje. Los padres además refieren que estos síntomas no han generado limitaciones para la realización de las actividades diarias.
Al examen físico, se trata de una paciente con signos vitales normales, talla de 48 cm (percentil 75 para la edad), normocéfala, con facies toscas, boca gruesa, puente nasal plano, raíz nasal rota, cuello ancho y corto, sin hallazgos a la auscultación cardiopulmonar, manos en garra, abdomen globoso con hepatoesplenomegalia, hernia umbilical reductible y cifosis dorso lumbar (Ver figura 1). Al examen neurológico, interacciona con el evaluador, sin embargo, no acataba órdenes ni emitía lenguaje, fuerza y sensibilidad conservadas.
Dentro de los estudios solicitados, la paciente cuenta con evaluación cardiovascular dentro de límites normales; ecografía abdominal con evidencia de aumento del tamaño del bazo en forma difusa, con medida de 8.4 cm en sentido longitudinal e hígado con aumento de tamaño y ecogenicidad en lóbulo derecho de 11.4 cm en sentido longitudinal; tomografía axial computarizada de senos paranasales con evidencia de hiperplasia adenoidea; resonancia magnética de columna dorsal con curva cifótica en unión dorso lumbar y presencia de imagen de aparente hemivértebra correspondiente a L1.
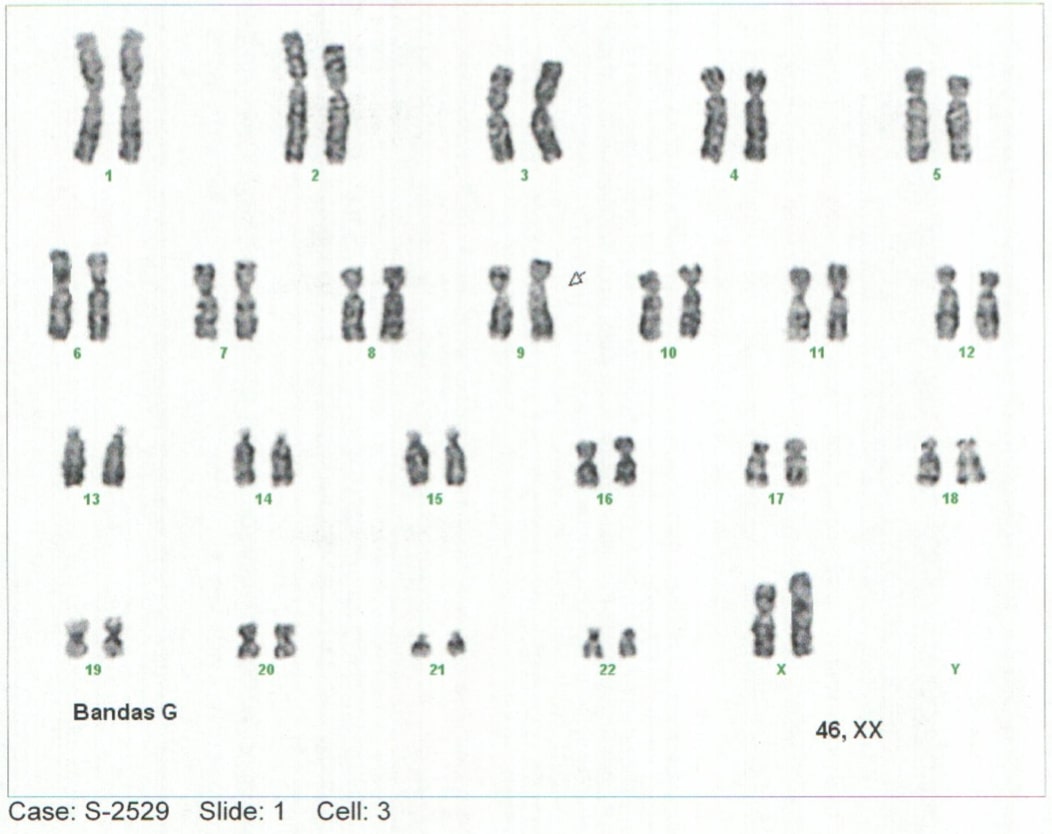
Dadas las características fenotípicas y los hallazgos paraclínicos hubo la sospecha de que se tratase de una enfermedad genética. Se procedió a la realización de cariotipo a partir de una muestra de sangre venosa periférica, documentando una complemento cromosómica 46 XX, 9qh+, correspondiente a un cariotipo femenino normal, con aumento de la región heterocromática del brazo largo del cromosoma 9 (polimorfismo benigno) (Ver figura 2).
Posteriormente, se evaluó la actividad de la enzima alfa-Liduronidasa obteniendo un valor de 0.04 nmol/ml (con valor de referencia normal: 2.02 - 16.1 nmol/ml). Se documentó entonces, actividad disminuida de dicha enzima, en un rango compatible con el diagnóstico Mucopolisacaridosis tipo I y en contexto de un cuadro clínico con manifestaciones severas y edad temprana de inicio de la patología, se realizó diagnóstico adicional de Síndrome de Hurler.
Figura 1. Características fenotípicas de la paciente.
Figura 2. Cariotipo (Muestra de sangre periférica del paciente).
Obsérvese un aumento heterocromático del brazo largo del cromosoma 9 (Polimorfismo normal).
Discusión
Se reportan los hallazgos clínicos y enzimáticos de una paciente con MPS I con variante síndrome de Hurler, la cual al requerir múltiples estudios y análisis de características clínicas se considera una enfermedad subdiagnosticada por lo que es importante conocer de su existencia, diagnóstico y tratamiento adecuado.
Existen más de 70 enfermedades de depósito lisosomal que se originan de alteraciones genéticas y consecuente falla enzimática, siendo la mucopolisacaridosis (MPS) uno de estos grupos (9). Dentro de este grupo se identificó a la MPS tipo I, la cual corresponde al diagnóstico de la paciente en mención y cuyos hallazgos fenotípicos típicos son dados por la deficiencia de la enzima alfa-L-iduronidasa, produciendo déficit en la degradación de Heparán Sulfato y Dermatán Sulfato, por tanto, su acumulación secundaria y progresiva en diversos tejidos tiene alta posibilidad de generar disfunción multiorgánica (2,4).
No hay evidencia en la literatura que establezca una epidemiología y comportamiento claro de esta enfermedad ni de los diferentes tipos que existen en Venezuela, país de origen de la paciente. Se conoce como promedio de prevalencia entre 1.04 a 4.8 por cada 100 000 nacidos vivos, con registro en países de Europa del Norte, donde es más frecuente la MPS tipo I. El país más común con reporte de dicha enfermedad es Noruega, donde existe aproximadamente 60 % de MPS I y prevalencia de 1.85 por cada 100 000 nacidos vivos.
Por otro lado, en Estados Unidos existe una prevalencia de 1.2 por cada 100.000 nacidos vivos de MPS II, así como en Japón y otros países de Asia es de mayor prevalencia el MPS II (53- 58 %) en relación a los tipos de mutaciones encontradas (5).
Dado el número de casos documentados en los países anteriormente mencionados, es de gran importancia reconocer las diversas mutaciones presentes en cada población; en países como Noruega, Dinamarca y Suiza es mayor el MPS I por dos mutaciones muy frecuentes: p.W402X y p.Q70X (5,8,10).
Las manifestaciones clínicas características son secundarias al almacenamiento progresivo de GAG que explican la hepatoesplenomegalia y las otras características clínicas que presenta la paciente, las cuales se puede evidenciar en los primeros meses de vida como lo son: disostosis múltiple o macroglosia, otitis media, obstrucción nasal, desordenes respiratorios, pérdida auditiva (11,12) Además, dichos pacientes pueden presentar alteraciones fenotípicas como facies toscas, retraso en el crecimiento, hernia umbilical, alteraciones cognitivas, así como musculoesqueléticas, cardiovasculares como cardiomiopatías agudas u oftalmológicas como opacidad corneal, entre otras (3,5,6,8,13).
Es importante resaltar que existen diferencias de mayor importancia en el síndrome de Hurler con respecto a otros síndromes (12). La principal es el deterioro neurológico cognitivo que inicia en promedio a los nueve meses, como es evidenciado en la paciente reportada, lo cual dificulta en un inicio su diagnóstico. Adicionalmente, también pueden coexistir otras alteraciones del neurodesarrollo donde se encuentran los trastornos motores, con retraso en los objetivos de acuerdo con lo esperado; así como retraso en comunicación receptiva y expresiva y el desarrollo de hipotonía (14).
La acumulación de GAG intravacuolares en el citoplasma de células endoteliales, miocitos y fibroblastos del tejido cardíaco, produce el continuo engrosamiento cardiaco que lleva a deformación valvular y conduce a regurgitación o estenosis (15). Se estima que alrededor del 98.2 % de los pacientes a los seis meses presentan hallazgos como alargamiento de circunferencia cefálica, dificultades en la alimentación, alopecia, organomegalia, alteraciones cardiacas, restricción articular o cifosis, entre otras ya mencionadas, hallazgos que la paciente ya presentaba para esta edad (14).
Ante la sospecha clínica de MPS se indican diversos estudios radiológicos dentro de los cuales se encuentran las radiografías de tórax, columna, pelvis y manos para detectar disostosis múltiple. Además, es importante realizar la medición de GAG en orina ya que al ser evaluaciones preliminares pueden ser útiles, aunque se ha encontrado alta incidencia de falsos positivos o negativos. Existe preferencia de pruebas cuantitativas de un solo GAG por medio de espectrometría de masas en tándem, aunque ninguno de estos estudios son diagnóstico definitivo de este grupo de enfermedades (6).
El diagnóstico definitivo de un paciente con hallazgos patognomónicos de MPS, así como evidencia paraclínica preliminar, se debe realiza por medio de medición enzimática, indicando como prueba positiva su deficiencia (16). Las pruebas enzimáticas se pueden realizar en todos los tipos de MPS, aunque algunos tipos (MPS II, III, IV, VI) tienen mayor dificultad para realizar debido a su sustrato por ser radiactivo (6,7).
En esta paciente el diagnóstico no se realizó de manera temprana como se documenta en la literatura, la paciente tenía 5 años y 5 meses para ese entonces, comparado con lo reportado en cohortes de pacientes con Mucopolisacaridosis tipo I y Síndrome de Hurler, que en promedio se diagnostica entre los 9 meses a 2 años (7,14); aunque se ha evidenciado al menos inicio de sintomatología al mes de nacimiento en 86 % (15).
Es común el retraso en el proceso diagnóstico debido a sintomatología inespecífica inicial y la poca correlación entre los hallazgos, sin embargo, el inicio de la sintomatología descrita, así como la severidad de esta orientó la sospecha diagnóstica.
El uso de diagnóstico molecular en pacientes con evidencia clínica y hallazgos de deficiencia enzimática o en portadores conocidos es poco utilizado por su costo y relevancia terapéutica (13,17); aunque esta propuesta difiere en otras fuentes bibliográficas ya que se indica uso de prueba moleculares para reconocer la alteración dentro de las diversas mutaciones que existen y así determinar su importancia epidemiológica (3,5,7,18).
La variabilidad fenotípica genera un desafío diagnóstico, sobre todo en pacientes con patrón leve de la enfermedad. Además de las alteraciones fenotípicas descritas en el caso expuesto, la resonancia nuclear de columna dorsal evidenció hemivértebra L1 con curva cifótica en unión dorsolumbar y ecografía de abdomen con hepatoesplenomegalia, correspondientes a consecuencia del depósito de GAG. Por lo cual, la correlación de los hallazgos en la literatura con respecto al caso presentado permite identificar su compatibilidad diagnóstica, como la realización del estudio enzimático, que identificó la disminución de la actividad de la enzima alfa-L-iduronidasa.
Por otro lado, las pruebas de tamizaje prenatal disponibles para esta enfermedad son pocas. El tamizaje por medio de análisis de GAG prenatal en líquido amniótico es poco fiable, y el uso de estudios moleculares en pacientes portadores conocidos es de gran costo (6,13,17). En Estados Unidos se realiza tamizaje de MPS I en neonatos, actualmente en cuatro estados (Illinois, Missouri, Kentucky y Pensilvania) midiendo la actividad enzimática de la alfa-L-iduronidasa; estrategia adaptada de programas de Taiwán e Italia (5).
Existen diferentes estrategias para el seguimiento y tratamiento, dentro de ellas está el seguimiento multidisciplinario de Neuropediatría, Pediatría, Fisioterapia, Ortopedia, Oftalmología, entre otros (3,19,20). Inicialmente, el trasplante de células madre hematopoyéticas representó una mejoría de efectos de depósito en MPS I, II, VI, dentro de los cuales se encuentra mejoría de la rigidez articular, hepatoesplenomegalia y retraso del crecimiento (19). Pese a lo anterior, no se ha evidenciado una mejoría significativa neurológica en los pacientes con MPS I , II y III, cuando estos han presentado alteraciones previo al tratamiento. Por otro lado, el uso de trasplante de cordón umbilical continúa siendo el estándar de oro en pacientes con MPS IH y II antes de los 2 a 2.5 años (3,6,20).
El uso de terapia de reemplazo enzimático en pacientes con MPS I, inicialmente publicado en 2001 empleando alfa-L-iduronidasa recombinante, permite la disminución de hepatoesplenomegalia, mejora la apnea del sueño, así como el crecimiento y movimiento articular, sin embargo, dicha enzima no puede superar la barrera hematoencefálica por lo que no puede evitar el deterioro neurológico (17,20–22). Se ha reservado en pacientes con afectación leve neurológica y en pacientes que van a ser tratados con trasplante de cordón (3,6,23). Existe poca evidencia de la efectividad a largo plazo y poca información bibliográfica del uso de esta terapia enzimática en pacientes con fenotipos leves, así como sus efectos en la historia natural de la enfermedad (24,25).
Los pacientes con enfermedades de depósito, en especial MPS I, con factores que determinan un deterioro progresivo requieren un manejo integral desde el momento del diagnóstico, al ser una patología donde la expectativa de vida no supera la segunda década de vida (6,17). No obstante, el uso de terapias como trasplante de cordón en pacientes con MPS IH han tenido mayor expectativa de vida en comparación a los que no han recibido ningún tratamiento (25).
Al ser una enfermedad infrecuente, se ha reportado poco en la literatura complicaciones diferentes a las manifestaciones clínicas (5,17), aunque está establecido el riesgo de complicaciones por alteraciones en la vía respiratorias que conlleva a infección u obstrucción, así como alteraciones cardiovasculares, dichas entidades tienden a ser las principales causas de muerte (6,11,12,17).
Por último, es importante resaltar el impacto social en las familiares de pacientes diagnosticados con MPS, por ello el adecuado acompañamiento de personal capacitado para brindar claridad y apoyo en las diversas etapas de la enfermedad. En dicho caso se demuestra el efecto que causa en la paciente tener un sistema de salud precario, donde la accesibilidad a terapia enzimática puede llegar a ser complejo por lo cual no ha sido fácil su administración.
Conclusiones
Por medio de avances en el abordaje temprano de pacientes con enfermedades de depósito como la mucopolisacaridosis, en específico la MPS tipo I, variante síndrome de Hurler (IH) permite generar un mejor desenlace y calidad de vida dentro de su expectativa de vida. Además de conocer los hallazgos clínicos sugestivos de esta enfermedad y el adecuado uso e interpretación de las pruebas a disposición para la identificación de este grupo de enfermedades, así como la búsqueda de nuevos tratamientos al entender la historia natural de la misma.
Conflicto de interés
Los autores declaran no existió conflicto de interés en la elaboración del manuscrito.
Referencias
1. Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008;3(1):1–7.
2. Filocamo M, Tomanin R, Bertola F, Morrone A. Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know. Ital J Pediatr. 2018;44(Suppl 2):129.
3. Feillet F, Wiedemann A, Jeannesson E, Jaussaud R, Journeau P. Mucopolisacaridosis. EMC - Pediatría. 2016;51(3):1–14.
4. Tebani A, Zanoutene-Cheriet L, Adjtoutah Z, Abily-Donval L, Brasse-Lagnel C, Laquerrière A, et al. Clinical and molecular characterization of patients with mucopolysaccharidosis type I in an Algerian series. Int J Mol Sci. 2016;17(5).
5. Khan SA, Peracha H, Ballhausen D, Wiesbauer A, Gautschi M, Mason RW, et al. Molecular Genetics and Metabolism. HHS Public Access Author. 2018;121(3):227–40.
6. Spranger WJ, Kliegman R. Nelson. Tratado de pediatria - Mucopolisacaridosis [Internet]. 20th Editi. Nelson. Tratado de pediatría. Elsevier España S.L.U.; 2016. 772–779 p.
7. Simon Jones, MD, Robert Wynn M. Clinical features and diagnosis. UpToDate.
8. Lee-Chen GJ, Wang TR. Mucopolysaccharidosis type I: Identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J Med Genet. 1997;34(11):939–41.
9. Yang C, Pan J, Linpeng S, Li Z, Tan H, Wu L. Identification of five novel mutations causing rare lysosomal storage diseases. Med Sci Monit. 2019;25:7634–44.
10. Scott HS, Bunge S, Gal A, Clarke LA, Phillip Morris C, Hopwood JJ, et al. Molecular Genetics of Mucopolysaccharidosis Type I: Diagnostic, Clinical, and Biological Implications. Hum Mutat. 1995;6:288302.
11. Bianchi PM, Gaini R, Vitale S. ENT and mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):127.
12. Suarez-Guerrero JL, Gómez Higuera PJI, Arias Flórez JS, Contreras-García GA. Mucopolisacaridosis: características clínicas, diagnóstico y de manejo. Rev Chil Pediatr. 2016;87(4):295–304.
13. Clarke LA, Atherton AM, Burton BK, Day-Salvatore DL, Kaplan P, Leslie ND, et al. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J Pediatr [Internet]. 2017;182:363–70. Available from: http://dx.doi.org/10.1016/j.jpeds.2016.11.036.
14. Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12(1):1–10.
15. Harmatz PR, Shediac R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front Biosci - Landmark. 2017;22(3):385–406.
16. Coutinho MF, Encarnação M, Matos L, Silva L, Ribeiro D, Santos JI, et al. Molecular characterization of a novel splicing mutation underlying mucopolysaccharidosis (MPS) type VI—Indirect proof of principle on its pathogenicity. Diagnostics. 2020;10(2):1– 11.
17. Peck DS, Lacey JM, White AL, Pino G, Studinski AL, Fisher R, et al. Incorporation of second-tier biomarker testing improves the specificity of newborn screening for mucopolysaccharidosis type i. Int J Neonatal Screen. 2020;6(1):1–10.
18. Shafaat M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic testing of Mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: in silico analysis of novel mutations. Metab Brain Dis. 2019;34(5):1447–55.
19. Li X, Xiao R, Chen B, Yang G, Zhang X, Fu Z, et al. A novel mutation of SGSH and clinical features analysis of mucopolysaccharidosis type IIIA. Med (United States). 2018;97(52).
20. Parini R, Deodato F, Di Rocco M, Lanino E, Locatelli F, Messina C, et al. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12(1):1–9.
21. Sawamoto K, Chen HH, Alméciga-Díaz CJ, Mason RW, Tomatsu S. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018;123(2):59–68.
22. Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019;2019(6).
23. Fecarotta S, Gasperini S, Parenti G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. Ital J Pediatr. 2018;44(Suppl 2):124.
24. Kuiper GA, Nijmeijer SCM, Roelofs MJM, van der Lee JH, Hollak CEM, Bosch AM. Limited data to evaluate real-world effectiveness of enzyme replacement therapy for mucopolysaccharidosis type I. J Inherit Metab Dis. 2019;42(5):762–75.
25. Eisengart JB, Rudser KD, Xue Y, Orchard P, Miller W, Lund T, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423–9.